疾病概述
 生長激素缺乏性侏儒症
生長激素缺乏性侏儒症故無論是下丘腦或垂體周圍病變致GHRH-GH生成或輸出障礙(如垂體性侏儒症),或GH受體缺陷(如Laron侏儒症)或周圍組織對IGF不敏感(如非洲侏儒)均可致生長遲緩,身材矮小見表。生長激素缺乏性侏儒症(growthhormonedeficiencydwarfism,GHD)又稱垂體性侏儒症(pituitarydwarfism),是指自嬰兒期或兒童期起病的腺垂體生長激素缺乏而導致生長發育障礙。其病因可為特發性或繼發性;可由於垂體病變(垂體性)或下丘腦和(或)垂體柄病變導致生長激素缺乏(下丘腦性);可為單一生長激素缺乏,也可伴腺垂體其他激素缺乏。GHD多見於男性。
症狀體徵
 體徵
體徵患者至青春期,性器官不發育,第二性徵缺如。男友生殖器小,與幼兒相似,睪丸細小,多伴有隱睪症,無鬍鬚;女性表現為原發性米徑,乳房不發育。單一性生長激素缺乏者可出現性器官發育與第二性徵,但往往明顯延遲。
智力發育一般正常,學習成績與同年齡者五差別,但年長後常因身材矮小而抑鬱寡歡,不合群,有自卑感。
繼發性生長激素缺乏性侏儒症除上述表現外,可伴有運發並的各種症狀,由下丘腦—垂體部位腫瘤引起者,可出現勢力減退,視野缺損,後期可出現顱內壓增高的表現,以及嗜睡、抽搐等。
疾病病因
 生長激素分子
生長激素分子病理生理
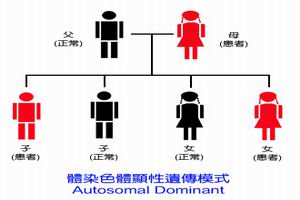 基因模式
基因模式生長激素可為單一性缺乏,但常伴有促性腺激素缺乏,有時也可伴有促甲狀腺激素或(和)促腎上腺皮質激素缺乏,後兩者往往無明顯臨床表現,常由實驗室檢查發現。
少數病人有家族史,其發病可能與遺傳有關,由於生長激素基因缺失所致。
生長激素缺乏性侏儒症也可繼發於下丘腦—垂體腫瘤,最常見者為顱咽管瘤,其他如感染(腦炎、腦膜炎)、創傷(圍生期腦損傷)、放射等均可影響腺垂體—下丘腦功能,引起繼發性生長激素缺乏性侏儒症。
少數患者血漿生長激素並不降低,甚至或升高,但生長介素濃度降低,注射生長激素後也不升高,提示肝細胞生長激素受體缺乏或有缺陷,對生長激素不敏感,由此引起的侏儒症稱為Laron侏儒症,多見於猶太人;也有周圍組織對生長介素不敏感,見於非洲侏儒。
診斷檢查
 檢查
檢查生長激素缺乏性侏儒症確診後,尚須進一步尋找致病原因。應作視野檢查、蝶鞍X線攝片等,必要時可作CT、磁共振等以除外垂體瘤。特發性者臨床上無明顯原因可找到。
治療方案
 生長激素缺乏性侏儒症
生長激素缺乏性侏儒症二、同化激素睪酮有促進蛋白質合成作用,對生長激素缺乏性侏儒症雖能於使用初期身高增加,但因同時可促進骨骺提早融合省長停止,患者最終身材仍然明顯矮小,療效很不理想,人工合成的同化類固醇有較強的的促進蛋白質合成作用而雄激素作用較弱,故可促進省長,並可減輕骨骺融合等副作用。臨床上常用者為苯丙酸諾龍,一般可在12歲後小劑量間歇套用,每周一次,每次10—12.5mg,肌肉注射,療程以一年為宜,有時第一年內可長高10cm左右,但以後生長減慢,身材仍矮小。
三、人絨毛膜粗性腺素能促使黃體的形成與分泌,或促進睪丸間質細胞分泌睪酮,只使用與年齡已達青春發育期、經上述治療身高不在增長者,每次500—1000U,肌肉注射,每周1—3次,每2—3個月為一療程,間歇2—3個月,可反覆套用1—2年,過早套用可引起骨骺融合,影響生長,於男孩可引起乳腺發育。繼發性生長激素缺乏性侏儒症應針對原發病進行治療。
預後及預防
 預防措施
預防措施1.若該病不能及時診斷和治療,將會導致成年後身材顯著矮小、心血管疾病發生率升高,而且有相當多病例伴有性腺發育不良、中樞性甲狀腺功能低下、促腎上腺皮質激素(ACTH)缺乏症。因此,不能得到醫治的生長激素缺乏性侏儒症(GHD)將會嚴重地影響今後的工作、學習、婚姻、心理和生活質量等,如能得到早期治療,可以將身高達到正常人高度範圍內。另外,對維持肌肉活力、改善心臟功能、延緩衰老、防治骨質疏鬆、治療肥胖等也起著重要作用。
2.下丘腦-垂體部位腫瘤引起者,可出現視力減退,視野缺損,後期可出現顱內壓力增高的表現,以及嗜唾、抽搐。
 生長激素缺乏性侏儒症
生長激素缺乏性侏儒症1.GH缺乏症有明顯的家族遺傳特點,可以做染色體檢查。
2.定期做好圍產期保健,避免圍生期病變史如:難產、宮內窒息等,以免造成腦部受損。
流行病學
生長激素缺乏性侏儒症是兒科內分泌疾病中比較常見的疾病,根據北京協和醫院1987年調查研究結果顯示:北京城市學齡兒童及青少年中,生長激素缺乏性侏儒症(IGHD)發病率為1/8644。國外報導IGHD約占全部矮小症患者的50%,我國廣大農村地區受衛生條件的限制,估計發病率高於城市。
