發病機制
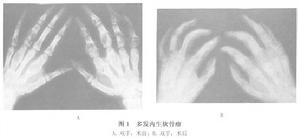 多發內生軟骨瘤病
多發內生軟骨瘤病腫瘤組織為白色,略有光澤,質脆呈半透明狀。摻雜黃色鈣化或骨化區,或有黏液樣退變區。顯微鏡下見分葉狀透明軟骨軟骨細胞成堆,有雙核者,單核大小均勻,染色不深。
臨床表現
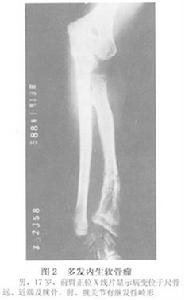 多發內生軟骨瘤病
多發內生軟骨瘤病通常發病年齡為10歲以內患兒,男性多於女性。
症狀與體徵:表現為可觸及的腫塊,但很少有疼痛腫瘤侵及手或足,由於多發病變可以造成病殘。病變侵及長管狀骨,使內生軟骨骨化不能正常進行骨骺板不能正常生長因而肢體可以出現短縮彎曲畸形。如前臂向尺側彎曲畸形,下肢膝外翻等。當患者達到成年時腫瘤可停止生長。成人多發內生軟骨瘤病可發生惡性變,惡變率約為5%~25%。
多發內生軟骨瘤病,常在兒童時期出現症狀,至青春期畸形明顯,以後逐漸穩定。
檢查
X線檢查:多發內生軟骨瘤病的每一個病變的X線表現與單發內生軟骨瘤相似,但為多發。且有骨骼畸形或短縮。其乾骺端可以增寬。
治療
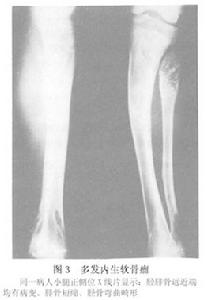 多發內生軟骨瘤病
多發內生軟骨瘤病由於病變的多發性,難以將每個內生軟骨瘤均予治療,對無症狀者可以不予治療但應隨診觀察對有症狀的具體部位,可以刮除病灶並植骨,對明顯的肢體畸形可以做截骨糾正。
多發內生軟骨瘤病有人報告可以隨著生長病變可以縮小甚至完全消失,而被正常組織所代替。但另一方面這種病變潛在性惡性改變的可能性較大,可變為軟骨肉瘤或骨肉瘤,如有惡性變發生,則應採取較徹底手術方法予以切除,甚至截肢。
預後
多發內生軟骨瘤具有潛在惡性變化的可能,惡變為軟骨肉瘤或骨肉瘤,應引起注意。
