
概述
 催化實驗
催化實驗催化即通過催化劑改變反應物的活化能,改變反應物的化學反應速率,反應前後催化劑的量和質均不發生改變的反應。
化學反應物要想發生化學反應,必須使其化學鍵發生改變,改變或者斷裂化學鍵需要一定的能量支持,能使化學鍵發生改變所需要的最低能量閾值稱之為活化能,而催化劑通過降低化學反應物的活化能而使化學反應更易進行,且大大提高反應速率。
我們知道,在化學反應中能改變化學反應速度而不影響化學平衡的作用稱為催化。其中能夠改變化學反應速度而本身的組成和數量在反應前後均不發生改變的物質稱為催化劑。催化劑可以使化學反應速率增加或減慢,其中增加化學反應速率的催化劑為正催化劑,反之,則為負催化劑。催化作用幾乎涉及到化學反應的各個領域。
催化是利用催化劑改變化學反應速度的一種工藝。許多化學工業要利用催化作用來獲得需要的反應速度。催化也是一種化工單元過程,催化劑本身在反應中不會被消耗,但催化劑會改變反應速度,一催化劑亦可能參與複數的催化反應。正催化劑可加速反應;負催化劑或抑制劑則會與反應物反應進而降低化學反應。可提高催化劑活性的物質稱為促進劑;降低催化劑活性者則稱為催化毒。
相較於未催化的反應,同溫度的催化反應擁有較低的活化能。催化劑可以藉由結合反應物達到極化的效果,如酸催化劑之於羰基化合物的合成;催化劑也可產生非自然的反應中間物,如以四氧化鋨催化烯烴的雙羥基化中產生的鋨酸鹽酯;催化劑亦可造成反應物的裂解,如制氫時產生的單原子氫。
很多物質都可以做催化劑,在無機物反應中,通常利用酸、鹼、金屬或金屬化合物作為催化劑,在有機物反應中多用有性的蛋白質分子——酶作為催化劑,生物體內許多化學反應都依賴酶來進行的。
概念的誕生
 催化概念的誕生
催化概念的誕生古代時,人們就已利用酶釀酒、制醋;中世紀時,鍊金術士用硝石作催化劑以硫磺為原料製造硫酸;13世紀,人們發現用硫酸作催化劑能使乙醇變成乙醚。直到19世紀,產業革命有力地推動了科學技術的發展,人們陸續發現了大量的催化現象。
如上所述,催化劑作用是在生產發展的同時為人們由淺入深地認識到的。1835年,貝采里烏斯(Berzelius)首先總結了此前的30多年間發現的催化作用。
為了解釋這一現象,他首先採用了“催化”這一名詞,並提出催化劑是一種具有“催化力”的外加物質,在這種作用力影響下的反應叫催化反應,即物質的能力“以物質本身存在,而非起親和力,去喚醒在某一特定溫度下還在沉睡的親和力”。這是最早關於催化反應的理論。
其認識的深入
 認識的深入
認識的深入人們對於催化作用特點的認識過程是漫長的。在這一認識過程中,許多科學家都親自從事化學實驗並發現了許多催化反應。通過長期實踐,逐漸積累加深了認識。
1781年,帕明梯爾(Parmentler)用酸作催化劑,使澱粉水解。1812年,基爾霍夫(Kirchhoff)發現,如果有酸類存在,蔗糖的水解作用會進行得很快,反之則很緩慢。而在整個水解過程中,酸類並無什麼變化,它好像並不參加反應,只是加速了反應過程。同時,基爾霍夫還觀測到,澱粉在稀硫酸溶液中可以變化為葡萄糖。1817年,戴維(Davy)在實驗中發現鉑能促使醇蒸氣在空氣中氧化。1838年,德拉托和施萬分別都發現糖之所以能發酵成為酒精和二氧化碳,是由於一種微生物的存在。貝采里烏斯就此提出,在生物體中存在的那些由普通物質、植物汁液或者血而生成無數種化合物,可能都是由此種類似的有機體組成。後來,居內將這些有機催化劑稱為“酶”。
1850年,威廉米(Wilhelmy)通過研究酸在蔗糖水解中的作用規律,第一次成功地分析了化學反應速度的問題,從此開始了對化學動力學的定量研究。1862年,聖·吉爾和貝特羅在實驗中發現,如果按照分子比將醋酸乙脂與水混合,經過幾星期之後於進行觀測,發現醋酸乙脂已部分水解成為乙醇和醋酸。這一反應的速度隨時間處長而呈遞減趨勢。再將乙醇與酸混合,反應生成了醋酸乙脂,平衡後的比例相同。這一反應的速度同樣很慢。但是,當有無機酸存在時,上述兩個反應則可在幾小時內完成。這樣,無機酸作為一種催化劑可以促進兩個反應向任一方向進行的反應速度。
1884年前後,包括奧斯特瓦爾德(Ostward)在內的幾位化學家研究了各種酸對酯的水解作用以及蔗糖轉化等現象的酸鹼催化作用的解釋,他認為催化劑現象的本質,在於某些物質具有一種特彆強烈的使原本沒有它參加而速度很慢的反應加速的特殊性能。
1895年奧斯特瓦爾德提出的催化定義為:“催化劑是一種物質,其能改變某一反應的反應速率而不能改變該反應的能量因素。他說,任何物質,如果它不參加到化學反應的最終產物中去,只是改變這個反應的速度即稱為催化劑。另外,他通過總結大量的實驗結果,根據熱力學第二定律,提出了平衡的達成,不能改變平衡常數。
1905年,勒·羅西諾(LeRosignol)和哈伯(Haber)等人,根據化學熱力學的原理,研究計算了氫、氮和氨在各種溫度和壓力平衡情況後,利用各種催化劑的幫助,研究出從空氣中的氮合成氨的實驗方法。催化與可逆過程的關係也有研究,催化不影響平衡狀態概念得以確立。從催化與平衡的討論中可以得出結論:在任何可逆反應中催化劑可加速正反應,也可加速逆反應,比如加氫催化劑在逆反應脫氫中也是有效的。
其理論的發展
 理論的發展
理論的發展在尋找催化劑和催化反應的過程的同時積累了大量的資料,使人們對催化劑和催化作用的認識不斷深入。關於催化反應的理論也逐步得以發展。催化劑為什麼能夠改變化學反應的速度,而它本身在反應後又不發生化學變化呢?為了解釋這一問題,在19世紀初期,就已經有人提出關於催化劑在反應中生成中間化合物的假說,認為催化劑之所以有所謂“催化能力”,是由於生成了中間化合物的結果。
1806年,德索爾姆(Desormes)和克雷蒙(Clement)研究一氧化氮對二氧化硫氧化的催化作用時,推想一氧化碳先與大氣中的氧反應生成某種中間化合物。這一中間化合物再與二氧化硫相互作用,此時把氧轉交給後者,中間物質自身又變為一氧化氮。一氧化氮可以再被空氣氧化,之後再把氧交給二氧化硫。如果按照這種概念,這種均相催化反應是交錯地進行的氧化還原過程的綜合。一個缺憾是他們沒有提出具體的反應的具體過程。
1806年,德索爾姆(Desormes)和克雷蒙(Clement)研究一氧化氮對二氧化硫氧化的催化作用時,推想一氧化碳先與大氣中的氧反應生成某種中間化合物。這一中間化合物再與二氧化硫相互作用,此時把氧轉交給後者,中間物質自身又變為一氧化氮。一氧化氮可以再被空氣氧化,之後再把氧交給二氧化硫。如果按照這種概念,這種均相催化反應是交錯地進行的氧化還原過程的綜合。一個缺憾是他們沒有提出具體的反應的具體過程。
1835年,貝采里烏斯提出的過程與克雷蒙和德索爾姆的概念最為類似,他認為催化反應由下列兩個過程交替進行:
2NO+O=N2O3
SO2+N2O3+H2O=H2SO4+2NO
可以看出,在貝采里烏斯所提出的過程中,三氧化氮就是相當於克雷蒙和德索爾姆所推想的把空氣中的氧轉交給二氧化硫的活性中間物質。在貝采里烏斯之後,威廉遜(Willianson)曾於1851年用相似的方法來解釋該反應的進行。從此,中間化合物這一概念得到確立,並在以後得到廣泛套用。
邢歇伍德(Hinshelwood)等人在1930年,以碘蒸氣為催化劑進行乙醛蒸氣的加熱分解反應,發現均相催化反應的速度常常與催化劑的深度成正比的。而在該反應中,作為催化劑的碘蒸氣的深度始終不變,邢歇伍德認為,這一事實說明由於催化劑K先與某一反應物A或B相互作用,生成了活性的中間化合物X,此中間化合物進一步轉變而生成C並使催化劑再生。他們用以下形式表達上述反應歷程:
A+K=X+……
X+B=C+K……
可見,活性的中間化合物的假說因此得以進一步的證實和完善,同時均相催化理論也得到了發展。
隨著更多實驗事實的發現和研究的不斷伸入,人們發現催化劑作用不僅是均相地進行,更多的是這一類反應則是在多相中進行。並且,這時反應物在相界面上的濃度更大,這種現象被稱為“吸附作用”。科學家們把吸附分為兩種類型,一種是簡單的物理吸附;另一種是吸附的同時形成化學鍵,稱為化學吸附,當然,這一類完成是吸附的同時形成化學鍵,稱為化學吸附,當然,這一類完成的過程是曲折的。
催化反應的吸附理論首先是由義大利人珀蘭尼(Polanyi)在1824年提出的。他認為,由於吸附作用使物質的質點相互接近,因而它們之間容易發生反應。他說,吸附作用是由於電力而產生的分子吸引力。
 理論的發展
理論的發展1834年,法拉第(Faraday)則提出了與上者不同的吸附理論,他認為催化反應不是電力使然,而是靠體物質相互吸收所產生的氣體張力。他認為,如果催化劑表面極為乾淨,氣體就會附著其上而凝結,一部分反應分子彼此接近到一定程度時,就會使新合力發生作用,抵消排斥力,因而使反應變得容易進行。朗繆爾(Langmuir)在1916年間,發表了一系列關於單分子表面膜的行為及性質,和關於固體表面吸附作用的研究成果影響到催化理論的形成。之後,科學界在1920年-1940年間大量的研究成果對催化吸附理論有著重大影響。
值得注意的是,在這一時期通過對吸附量和脫附速度的研究,以及關於催化過程中催化失去催化活力的研究,得出了對多相催化理論有著根本意義的結論,即催化反應是在催化劑表面直接相連的單分子層中進行的。
就此,美國人泰勒(Taylor)於1925年首先提出了活性中心理論,其出發點即催化劑失去活性這一實驗事實。他認為催化劑的表面是不均勻的,位於催化劑表面微型晶體的棱和頂角處的原子具有不飽合的鍵,因而形成了活性中心,催化反應只發生在這一活性中心。泰勒的理論很好地解釋了催化劑製備對活性的影響以及毒物對活性的作用。
在泰勒之後,前蘇聯的兩位科學家對活性中心理論進行了進一步的完善和發展。1929年,巴蘭金(Баландин)提出了多位催化理論,認為催化劑活性中心的結構應當與反應物分子在催化反應過程中發生變化的那部分結構處於向何對應。這一理論把催化活化看作反應物中的多位體的反應過程,並且這個作用會引起反應物中價鍵的變形,並使反應物分子活化,促成新價鍵的形成。另一位蘇聯人柯巴捷夫(н.И.Koбазeв)於1939年提出了活性集團理論,與泰勒不同的是他認為活性中心是催化劑表面是上非晶體中幾個催化劑原子組成的集團。
20世紀50年代以後,隨著固體物理的發展,催化的電子理論應運而生。在這一層面上,科學家們得到了豐富的實驗成果,他們將金屬催化性質與基電子行為和甲子能級聯繫起來。70年代,根據催化劑表面的原子結構、絡合物中金屬原子簇的結構和性質,利用量子化學理論,對多相催化的高分散的金屬催化劑活性集團產生催化活性的根源進行研究。在科學突飛猛進的今天,催化作用的實質以及催化劑發生作用的秘密正逐漸為人們所認知。
二次世界大戰前化學工業基於煤炭工業,著重於合成氨、硝酸生產、油脂硬化及甲醇合成,20世紀50年代初期,石油化學工業的發展迅速,導致許多新燃料的面市、聚合物工業的突起、選擇氧化、氫甲醯化和其它均相反應也逐漸變得重要。20世紀70年代石油危機使人們更加珍惜能源和原材料的合理利用。汽車尾氣和工業排放污染治理受到重視。催化在這些方面起著決定作用。
約有80%的化學過程使用了催化劑,它的銷售額接近100億美元,但卻只有其所創造產品收入的1%的催化劑市場正以每年約10%的速度遞增。
相關種類
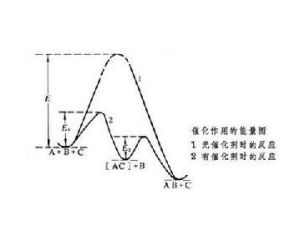 催化示意圖
催化示意圖均相催化
催化劑與反應物同處於一均勻物相中的催化作用。有液相和氣相均相催化。液態酸鹼催化劑,可溶性過渡金屬化合物催化劑和碘、一氧化氮等氣態分子催化劑的催化屬於這一類。均相催化劑的活性中心比較均一,選擇性較高,副反應較少,易於用光譜、波譜、同位素示蹤等方法來研究催化劑的作用,反應動力學一般不複雜。但均相催化劑有難以分離、回收和再生的缺點。
多相催化
多相催化發生在兩相的界面上,通常催化劑為多孔固體,反應物為液體或氣體。多相催化反應通常可按下述七步進行:①反應物的外擴散──反應物向催化劑外表面擴散;②反應物的內擴散──在催化劑外表面的反應物向催化劑孔內擴散;③反應物的化學吸附;④表面化學反應;⑤產物脫附;⑥產物內擴散;⑦產物外擴散。這一系列步驟中反應最慢的一步稱為速率控制步驟。化學吸附是最重要的步驟,化學吸附使反應物分子得到活化,降低了化學反應的活化能。因此,若要催化反應進行,必須至少有一種反應物分子在催化劑表面上發生化學吸附。固體催化劑表面是不均勻的,表面上只有一部分點對反應物分子起活化作用,這些點被稱為活性中心。
生物催化
酶是一種生物催化劑,生物體內的所有化學變化幾乎都是在酶催化下進行的,酶的催化作用稱為生物催化。酶的催化活性高,選擇性強。生物催化在常溫中性條件下進行,高溫、強酸和強鹼都會使酶喪失活性。離體的酶仍具有催化活性,可製成各種酶製劑套用在醫學和工農業生產上。其他還有電催化、光助催化、光電催化等。按催化作用機理,可把催化劑分為金屬催化劑、金屬氧化物催化劑、配位(絡合)催化劑、酸鹼催化劑和多功能催化劑。
金屬催化
金屬催化劑主要用於脫氫和加氫反應。有些金屬還具有氧化和重整的催化活性。金屬催化劑主要是指4、5、6周期的某些過渡金屬,如鐵、金、鉑、鈀、銠、銥等。金屬催化主要決定於金屬原子的電子結構,特別是沒有參與金屬鍵的d軌道電子和d空軌道與被吸附分子形成吸附鍵的能力。因此,金屬催化劑的化學吸附能力和d軌道百分數是決定催化活性的主要因素。
金屬氧化物催化
主要是指過渡金屬氧化物催化,非過渡金屬氧化物催化已歸入酸鹼催化。過渡金屬氧化物催化劑廣泛用於氧化、加氫、脫氫、聚合、加合等反應。實用的金屬氧化物催化劑常為多組分氧化物的混合物,很多金屬氧化物催化劑是半導體,其化學組成大多是非化學計量的,因此,催化劑組分很複雜。金屬氧化物催化劑的導電性和逸出功、金屬離子的d電子組態、氧化物中晶格氧特性、半導體電子能帶、催化劑表面吸附能力等,都與催化劑的催化活性有關。
配位(絡合)催化
金屬、特別是過渡金屬及其化合物有很強的絡合能力,能形成多種類型的絡合物。某些分子與金屬(或金屬離子)絡合後便易於進行某特定反應,該反應稱為配位(絡合)催化反應,該金屬或其化合物起絡合催化劑作用。過渡金屬絡合催化劑在溶液中作為均相催化劑方面的研究和套用較多。過渡金屬絡合催化作用一般都是配位(絡合)催化作用,即催化劑在其空配位上絡合活化反應物分子。絡合催化劑一般都是金屬絡合物或化合物,如鈀、銠、鈦、鈷的絡合物等。
酸鹼催化
阿倫尼烏斯酸鹼、布侖斯惕酸鹼、路易斯酸鹼(見酸鹼理論)的催化作用都屬酸鹼催化作用。酸鹼催化可分均相催化和多相催化。許多離子型有機反應,如水解、水合、脫水、縮合、酯化、重排等,常可用酸鹼均相催化。固體酸催化劑廣泛用於催化裂化、異構化、烷基化、脫水、氫轉移、歧化、聚合等反應。
多功能催化
若反應物A直接變成產物B的反應難以進行,則可通過幾個催化化學反應來實現。例如SⅠ、SⅡ分別為兩個反應的催化劑,則可將SⅠ、SⅡ混合起來製成雙功能催化劑,使A→B的反應實現。這就是多功能催化。多功能催化與一個催化反應的多步驟是有區別的,此處的C不是在催化劑表面形成的中間絡合物,而是由SⅠ表面脫附出來的、有其自己的結構和熱力學性質的化學物質。套用現代化學工業的巨大成就與催化劑的使用是分不開的。約80%以上的化學工業產品是藉助於催化過程來生產的,可以說,沒有催化劑就沒有現代化學工業。
其原理
 催化原理
催化原理降低活化能
在催化反應過程中,至少必須有一種反應物分子與催化劑發生了某種形式的化學作用。由於催化劑的介入,化學反應改變了進行途徑,而新的反應途徑需要的活化能較低,這就是催化得以提高化學反應速率的原因。例如,化學反應A+B─→AB,所需活化能為E,在催化劑C參與下,反應按以下兩步進行:
A+C─→AC,所需活化能為E1
AC+B─→AB+C,所需活化能為E2
E1、E2都小於E(見圖)。催化劑C只是暫時介入了化學反應,反應結束後,催化劑C即行再生。
按阿倫尼烏斯方程k=Ae-E/RT(式中A為指前因子;R為氣體常數;T為熱力學溫度),以反應速率常數k表示的反應速率主要決定於反應活化能E,若催化使反應活化能降低ΔE,則反應速率即提高e-ΔE/RT倍。催化反應一般能降低活化能約10千卡/摩爾,若反應在300K下進行,則反應速率可增加約1.7×10倍。
催化劑表面性能
多相催化是工業上套用最多的,這種催化作用在催化劑表面上進行,因此,固體催化劑的表面性質對催化作用就會有很大影響。催化劑比表面積大,表面上活化中心點多,表面對反應物吸附能力強,這些都對催化活性有利,因為化學吸附能降低反應活化能。表面孔隙度大和孔徑大小合適對催化劑的選擇性有利,例如分子篩催化劑的極強的選擇性,就是由於它的孔徑尺寸只能允許某種分子進入孔內,到達催化劑表面而被催化。
催化劑的作用方式
催化劑與反應物分子怎樣發生化學作用,這與催化劑和反應物分子本身的性質有關。實驗證明:有機化合物的酸催化反應一般是通過正碳離子機理進行的;鹼催化反應則是由OH、RO、RCOO等陰離子起催化作用,例如:
CH3COOC2H5+OH─→CH3COOH+C2H5O
C2H5O+H2O─→C2H5OH+OH
過渡金屬化合物催化劑在均相催化反應中的作用是絡合催化作用;醇鈉催化丁二烯聚合是通過自由基機理進行的,例如:
C4H6+NaR─→R•+C4H6Na•
C4H6Na•+nC4H6─→Na(C4H6)n+1
催化劑的性能指標
①催化活性:催化劑參與了化學反應,降低了化學反應的活化能,大大加快了化學反應的速率。這說明催化劑具有催化活性。催化反應的速率是催化劑活性大小的衡量尺度。活性是評價催化劑好壞的最主要的指標。②選擇性:一種催化劑只對某一類反應具有明顯的加速作用,對其他反應則加速作用甚小,甚至沒有加速作用。這一性能就是催化劑選擇性。催化劑的選擇性決定了催化作用的定向性。可通過選擇不同的催化劑來控制或改變化學反應的方向。③壽命或穩定性:催化劑的穩定性以壽命表示。它包括熱穩定性、機械穩定性和抗毒穩定性。
工藝單元組成
 有機廢氣催化燃燒淨化設備
有機廢氣催化燃燒淨化設備催化燃燒的工藝組成不同的排放場合和不同的廢氣,有不同的工藝流程。但不論採取哪種工藝流程,都由如下工藝單元組成。
①廢氣預處理為了避免催化劑床層的堵塞和催化劑中毒,廢氣在進入床層之前必須進行預處理,以除去廢氣中的粉塵、液滴及催化劑的毒物。
②預熱裝置預熱裝置包括廢氣預熱裝置和催化劑燃燒器預熱裝置。因為催化劑都有一個催化活性溫度,對催化燃燒來說稱催化劑起燃溫度,必須使廢氣和床層的溫度達到起燃溫度才能進行催化燃燒,因此,必須設定預熱裝置。但對於排出的廢氣本身溫度就較高的場合,如漆包線、絕緣材料、烤漆等烘乾排氣,溫度可達300℃以上,則不必設定預熱裝置。預熱裝置加熱後的熱氣可採用換熱器和床層內布管的方式。預熱器的熱源可採用煙道氣或電加熱,目前採用電加熱較多。當催化反應開始後,可儘量以回收的反應熱來預熱廢氣。在反應熱較大的場合,還應設定廢熱回收裝置,以節約能源。預熱廢氣的熱源溫度一般都超過催化劑的活性溫度。為保護催化劑,加熱裝置應與催化燃燒裝置保持一定距離,這樣還能使廢氣溫度分布均勻。從需要預熱這一點出發,催化燃燒法最適用於連續排氣的淨化,若間歇排氣,不僅每次預熱需要耗能,反應熱也無法回收利用,會造成很大的能源浪費,在設計和選擇時應注意這一點。
③催化燃燒裝置一般採用固定床催化反應器。反應器的設計按規範進行,應便於操作,維修方便,便於裝卸催化劑。在進行催化燃燒的工藝設計時,應根據具體情況,對於處理氣量較大的場合,設計成分建式流程,即預熱器、反應器獨立裝設,其間用管道連線。對於處理氣量小的場合,可採用催化焚燒爐見圖16-13,把預熱與反應組合在一起,但要注意預熱段與反應段間的距離。
在有機物廢氣的催化燃燒中,所要處理的有機物廢氣在高溫下與空氣混合易引起爆炸,安全問題十分重要。因而,一方面必須控制有機物與空氣的混合比,使之在爆炸下限;另一方面,催化燃燒系統應設監測報警裝置和有防爆措施。
所需原料
 催化劑
催化劑助催化劑:本身不具有催化活性,但加入後(加入量一般低於催化劑量的10%)可顯著提高催化劑的活性、選擇性和穩定性。一種工業催化劑往往要加入幾種助催化劑,才能使催化劑的活性、選擇性和壽命都達到預定要求。例如,合成氨用的雙促進催化劑鐵-三氧化二鋁-氧化鉀中的三氧化二鋁和氧化鉀就是助催化劑。
抑制劑:催化劑中含有的能降低催化劑活性的物質。在多數場合,它是在催化劑製備過程中由原料不可避免地帶入的。在催化反應過程中,由反應物帶入的抑制劑則稱催化毒物。
催化劑載體:不具有催化活性的用來負載催化劑的固體物質。把催化劑成分分散負載在載體上製成的催化劑稱負載型催化劑。常用的催化劑載體有活性碳、硅藻土、活性氧化鋁、矽膠和分子篩。對載體的要求是機械強度高、熱穩定性和化學穩定性好。載體雖不具有催化活性,但它可能與催化劑發生化學作用,載體也改變了催化劑的表面性能,因而選用合適的載體,也可以提高催化劑的活性、選擇性和壽命。
由於有機物催化燃燒的催化劑分為貴金屬以鉑、鈀為主和賤金屬催化劑。貴金屬為活性組分的催化劑分為全金屬催化劑和以氧化鋁為載體的催化劑。全金屬催化劑是以鎳或鎳鉻合金為載體,將載體做成帶、片、丸、絲等形狀,採用化學鍍或電鍍的方法,將鉑、鈀等貴金屬沉積其上,然後做成便於裝卸的催化劑構件。
由氧化鋁作載體的貴金屬催化劑,一般是以陶瓷結構作為支架,在陶瓷結構上塗覆一層僅有0.13mm的α-氧化鋁薄層,而活性組分鉑、鈀就以微晶狀態沉積或分散在多孔的氧化鋁薄層中。
但由於貴金屬催化劑價格昂貴,資源少,多年來人們特別注重新型的、價格較為便宜的催化劑的開發研究,我國是世界上稀土資源最多的國家,我國的科技工作者研究開發了不少稀土催化劑,有些性能也較好。
相關提示
 催化劑
催化劑在催化劑使用過程中,由於體系中存在少量的雜質,可使催化劑的活性和選擇性減小或者消失,這種現象叫催化劑中毒。這些能使催化劑中毒的物質稱之為催化劑毒物,這些毒物在反應過程中或強吸附在活性中心上,或與活性中心起化學作用而變為別的物質,使活性中心失活。毒物通常是反應原料中帶來的雜質,或者是催化劑本身的某些雜質,另外,反應產物或副產物本身也可能對催化劑毒化,一般所指的是硫化物如H2S、硫氧化碳、RSH等及含氧化合物如H2O、CO2、O2以及含磷、砷、鹵素化合物、重金屬化合物等。
毒物不單單是對催化劑來說的,而且還針對這個催化劑所催化的反應,也就是說,對某一催化劑,只有聯繫到它所催化的反應時,才能清楚什麼物質是毒物。即使同一種催化劑,一種物質可能毒化某一反應而不影響另一反應。按毒物與催化劑表面作用的程度可分為暫時性中毒和永久性中毒。暫時性中毒亦稱可逆中毒,催化劑表面所吸附的毒物可用解吸的辦法驅逐,使催化劑恢復活性,然而這種可再生性一般也不能使催化劑恢復到中毒前的水平。永久性中毒稱不可逆中毒,這時,毒物與催化劑活性中心生成了結合力很強的物質,不能用一般方法將它去除或根本無法去除。
催化劑的老化主要是由於熱穩定性與機械穩定性決定的,例如低熔點活性組分的流失或升華,會大大降低催化劑的活性。催化劑的工作溫度對催化劑的老化影響很大,溫度選擇和控制不好,會使催化劑半熔或燒結,從而導致催化劑表面積的下降而降低活性。另外,內部雜質向表面的遷移,冷熱應力交替所造成的機械性粉末被氣流帶走。所有這些,都會加速催化劑的老化,而其中最主要的是溫度的影響,工作溫度越高,老化速度越快。因此,在催化劑的活性溫度範圍內選擇合適的反應溫度將有助於延長催化劑的壽命。但是,過低的反應溫度也是不可取的,會降低反應速率。
為了提高催化劑的熱穩定性,常常選擇合適的耐高溫的載體來提高活性組分的分散度,可防止其顆粒變大而燒結,例如以純銅作催化劑時,在200℃即失去活性,但如果採用共沉積法將Cu載於Cr2O3載體上,就能在較高的溫度下保持其活性。
生活中的自然現象
| 人的意識也是以自然方式發生的物質世界。人和人的意識是自然界發展的最高產物。物質世界具有系統性、複雜性和無窮多樣性。它既包括人類已知的、也包括人類未知的物質世界。讓我們一起走進自然吧~~ |
