 遺傳性口形紅細胞增多症
遺傳性口形紅細胞增多症遺傳性口形細胞增多症在世界各民族中均有發現。我國亦有少數病例報導。本病為遺傳性疾病,由常染色體顯性傳遞。男女均可得病。各家族間溶血的程度很不一致。在部分患者中,本病的基因與rh血型的基因位於同一染色體上。兩種基因不聯接時,則溶血較嚴重。對本病的病因和發病機理目前尚不清楚。初步認為本病的缺陷也在紅細胞膜的支架蛋白。各病例的分子異常已發現有多種類型,所涉及的支架蛋白組成可以不同,所涉及的收縮蛋白的分子區域可以不同,膜蛋白缺失的量可以不等。這種異常細胞的破壞主要也是在脾臟內。
本病與遺傳性球形細胞增多症有相似之處,如細胞形態異常者在脾臟內破壞增多,胞膜中存在著某種通透性方面的缺陷促使三磷酸腺苷的利用加速。但致使紅細胞變成口形的原因究竟是什麼,還有待科學家的進一步探討。
引起本病膜缺陷的分子遺傳學異常有:膜收縮蛋白僅鏈基因異常(稱為僅HE突變)、膜收縮蛋白僅鏈低表達等位基因(稱為膜收縮蛋白B鏈基因異常、蛋白異常及血型糖蛋白C和D(D為C的變異型)缺乏。其中主要為膜收縮蛋白結構異常,少數為紅細胞膜的蛋白缺乏或蛋白與錨蛋白的結合缺陷。
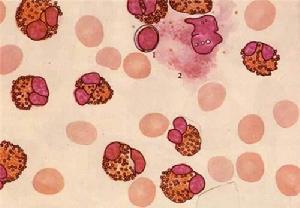
遺傳性口形紅細胞增多症可分遺傳性和獲得性,診斷主要依靠形態學。凡在外周血塗片中見到成熟紅細胞形狀呈卵圓形、口形、棒形或臘腸形,其橫經與縱經之比25%,並有家族遺傳史者可診斷為遺傳性口形紅細胞增多症。正常人的成熟紅細胞也可有少數(<5%)為口形,最多不應超過15%。
外周血中口形紅細胞超過25%,其臨床表現及血液學改變差異很大。
1無溶血(隱匿型):口形紅細胞雖增多,但無溶血表現。
2輕度溶血(溶血代償型):紅細胞壽命比正常稍短,網織紅細胞輕度增高,結合珠蛋白低於正常,由於造血功能的代償,多不出現貧血。絕大多數病人屬於這一類型。嚴重的可在新生兒期出現高膽紅素血症,甚至需要換血治療。合併感染時可出現骨髓不增生危象,亦有合併膽石症的報導。
1.有黃疸、貧血、脾大,多為幼年發病,可有家族史;
2.過度疲勞或感染可誘發溶血危象;
3.除一般溶血性貧血化驗所見;可見血片中有球形或口形紅細胞,數量可從1-2%到60-70%,多數在10%以上(正常人一般低於5%);
4.紅細胞滲透脆性增高;
5.酸化甘油溶解試驗陽性;
6.SDS-聚丙烯山胺凝膠電泳進行紅細胞膜蛋白分析,部分病例可發現膜骨架蛋白缺陷。
貧血程度的變化殊異。紅細胞計數一般為300萬~400萬/μl;在再生障礙危象時可降至100萬/μl以下,血紅蛋白亦成比例地下降.由於紅細胞呈球樣,而平均紅細胞體積正常,紅細胞的平均直徑稍低於正常,因而紅細胞酷似小球形紅細胞,平均紅細胞血紅蛋白的濃度增高。網織紅細胞增多(達15%~30%)與白細胞增多均為常見。
紅細胞的滲透脆性呈特徵性增高。但輕症者若不先在37℃下無菌的去纖維蛋白血中孵育24小時,試驗結果可能正常,直接抗球蛋白試驗(庫姆試驗)陰性。紅細胞自溶現象增加,但加入葡萄糖後可被糾正。
遺傳性口形紅細胞增多症沒有貧血或僅有輕度貧血者,一般不需要治療。溶血嚴重的,做脾切除術可使血紅蛋白和網織紅細胞恢復或接近正常。但脾切除後紅細胞形態異常變得更為明顯。由於嬰幼兒HE中一部分可自行減輕或緩解,脾切除術應在3歲以後考慮,的確需切脾者最好也在5歲以後進行。無症狀或僅有輕度貧血對身體健康影響不大者不需治療。如有較明顯溶血性貧血的應做脾切除。脾切除後口形紅細胞的特徵依然存在,但溶血可停止或減輕,血紅蛋白可恢復正常,又可防止長期慢性溶血導致的併發症,如膽石症等。
