概述
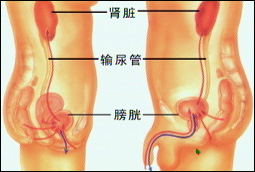 膜增生性腎小球腎炎
膜增生性腎小球腎炎流行病學
 膜增生性腎小球腎炎
膜增生性腎小球腎炎病因
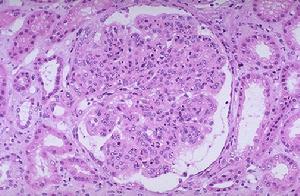 細胞圖
細胞圖原發性膜增生性腎炎病因不明,一般認為Ⅰ型為免疫複合物病;Ⅱ型為免疫複合物及自身抗體性疾病可能與遺傳有關。
繼發性膜增生性腎炎中混合性冷球蛋白血症有3種亞型。Ⅰ型冷球蛋白血症是單株峰球蛋白,通常為骨髓瘤蛋白。Ⅱ型通常為單株峰IgM球蛋白結合IgG,又稱抗IgG類風因子,而Ⅲ型則是多株峰免疫球蛋白。Ⅱ型和Ⅲ型冷球蛋白血症易發生腎損害。其病理特徵為系膜細胞大量增生、白細胞尤其單核細胞浸潤、腎小球基膜增厚有雙軌現象。約1/3病例有中小動脈炎,毛細血管內有微血栓形成MPGN的病因與發病機制
 膜增生性腎小球腎炎
膜增生性腎小球腎炎不十分明確Ⅰ型MPGN認為是免疫複合物病,由相對大的難溶的免疫複合物反覆持續沉積引起Ⅱ型MPGN患者血清中也存在免疫複合物,冷球蛋白補體異常、血清C3持續降低。均提示免疫複合物在Ⅱ型MPGN中的作用。Ⅱ型MPGN患者血清中可檢出C3腎炎因子(C3NeF),C3NeF是C3bBb轉化酶的自身抗體,使C3bBb作用加強導致補體旁路持續激活,產生持續低補體血症和基膜變性所以補體代謝障礙為中心環節。
另外,Ⅱ型MPGN腎移植中常復發,可能因病人血清中存在能引起異常糖蛋白形成的物質沉積於基底膜而導致腎炎。
本病可能與遺傳有關,Ⅱ型MPGN患者常出現HLA-B7大多Ⅰ型MPGN病人具有特殊B細胞同種抗原。
發病機制
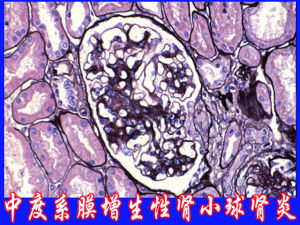 中度膜增生性腎小球腎炎
中度膜增生性腎小球腎炎根據各種免疫複合物沉積在腎小球基底膜內和系膜區的形式及沉積的程度不同將MPGN分為3種類型。
1.Ⅰ型 以內皮下及系膜區的複合物沉積為主。Ⅰ型與病毒、細菌及寄生蟲感染及一些免疫複合物疾病有關(如遺傳性補體缺失、SLE、混合性冷球蛋白血症、SBE、分流性腎炎、淋巴瘤、血吸蟲病),但常為特發性。在Ⅰ型MPGN患者中33%~50%出現低補體血症,25%~30%患者Clq、C4及C5降低15%~20%的患者B因子降低。
2.Ⅱ型 被稱為自身免疫性疾病在電鏡下可觀察到沿著基底膜的層黏蛋白層呈均勻一致的帶狀沉積,此型又稱為緻密物沉積病(DDD),還常伴有上皮下類似駝峰樣沉積物沉積PAS染色有時可見毛細血管襻上呈條帶狀深染Ⅱ型主要被認為與鏈球菌感染有關由於鏈球菌與腎臟抗原有交叉反應,可引起抗體介導的腎損害。Ⅱ型常並發血漿低C3水平因為部分患者血中存在補體激活物,一種自身抗體,也稱致腎炎因子或C3腎炎因子直接抗C3bBb,改變C3旁路轉化,通過與轉化酶結合,阻止一些正常抑制因子如H因子等作用,增加了補體的活化及消耗。C3腎炎因子在Ⅰ型和Ⅱ型MPGN中多見,尤其是在Ⅱ型中更為常見。部分與脂質營養不良有關。由於MPGNⅡ型主要是基底膜的損害,如有大量的緻密沉積物沉積在基底膜上,這些沉積物可以激活補體,補體被一些特殊物質如核糖酶激活,通常又可以激活旁路途徑,使C3腎炎因子繼發性持續增加而導致血中補體C3的下降。Ⅱ型MPGN中。70%的患者補體C3和B因子降低。
3.Ⅲ型 內皮下系膜區及上皮下都有沉積物沉積。Ⅲ型和Ⅰ型的區別在於上皮下是否有沉積。
伴隨時間的遷移MPGN的病理改變多從增生走向明顯的硬化亞型為局灶型MPGN時病變可移行為瀰漫經典型MPGN。部分小兒或青少年,開始為瀰漫性MPGN,亞型多為分葉型,之後可移行為局灶型或完全緩解。
病理變化
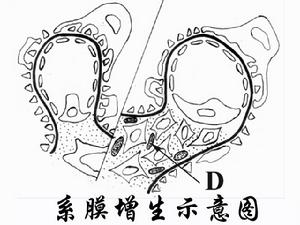
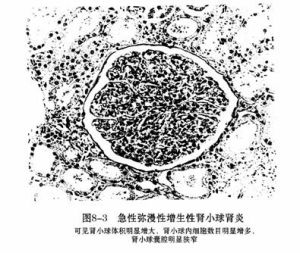
光鏡下,Ⅰ型典型的病變是,瀰漫性系膜細胞和系膜基質重度增生,沿內皮細胞和基底膜之間長入或插入(mesangialinterposition),使毛細血管壁增厚,鍍銀染色或PAS染色基底膜呈雙層(doublecontour)或多層狀改變,這是由於系膜基質插入造成的。由於系膜細胞和系膜基質重度增生,系膜區域擴大導致腎小球呈明顯的分葉狀。Ⅱ型膜增生性腎小球腎炎也呈現這些病變,但與Ⅰ型相比,系膜增生輕,腎小球分葉狀結構不明顯。
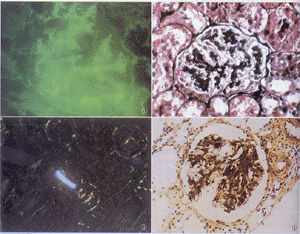
免疫螢光Ⅰ型膜增生性腎小球腎炎顯示IgG和補體C3呈顆粒狀和團塊狀沉積於毛細血管壁和系膜區。Ⅱ型膜增生性腎小球腎炎顯示大量補體C3沉積於毛細血管壁和系膜區。
電鏡下系膜細胞和系膜基質增生伴系膜插入,即增生的系膜細胞和系膜基質插入到基底膜和內皮細胞之間。系膜基質和基底膜形態相似,因而看上去似有兩層或多層基底膜,這就是光鏡下所見的雙層或多層的形態,一層是原有基底膜,另幾層是插入到內皮細胞和基底膜之間的系膜基質。腎小球內可見電子緻密物。Ⅰ型大量電子緻密物位於基底膜內皮側及系膜區域,少量見於上皮下;Ⅱ型電子緻密物的電子密度較Ⅰ型明顯高,沿基底膜緻密層呈帶狀分布。
臨床表現
 臨床表現
臨床表現本病發病時,至少有1/2的患者表現為腎病綜合徵;約1/4的患者表現為無症狀性血尿和蛋白尿;還有1/4~1/3的患者表現為急性腎炎綜合徵,伴有紅細胞及紅細胞管型尿、高血壓和腎功能不全。約有一半的患者可有前驅呼吸道感染史,40%在起病前有抗“O”滴度升高和鏈球菌感染的其他證據有的患者可發生部分脂質營養不良(Barraquar-Simmons病),尤其是Ⅱ型病變甚至可以在還沒有腎臟病臨床表現時發生。某些患者可顯示X-連鎖遺傳。先天性的補體和a1-抗胰蛋白酶缺乏也易發生在本病Ⅰ型。在腎病綜合徵時可發生腎靜脈血栓形成儘管本病發展有高度的個體差異性,但本病病情總體上呈緩慢的進行性進展。因本病Ⅰ型和Ⅱ型組織病理和免疫病理改變不一樣且為兩種類型的形態,臨床上多認為它們是代表不同的疾病。臨床上Ⅱ型更傾向於表現為腎炎徵象,新月體腎炎和急性腎衰的伴發率高,而Ⅰ型具有更多腎病的特徵,常有先驅感染和貧血;Ⅱ型患者血清常常有持續的低補體血症,並且發病年齡較小,幾乎所有患者發病均在20歲以下,儘管也有例外此外Ⅱ型更容易在腎移植後復發。
Ⅲ型很少見,主要發生在兒童和青年,10~20歲為高峰,<2歲,>40歲少見。男女發病接近。對於該型的臨床表現描述很少,基本與Ⅰ型的長期臨床改變相似。據Strife的描述Ⅲ型有C3水平降低,但無C3腎炎因子。非腎綜性蛋白尿的預後比腎病綜合徵表現者要好該型進入終末期腎病的個體差異比較大,在長期的病程中有些患者病情可以比較穩定甚至逐漸改善。
診斷
 膜增生性腎小球腎炎
膜增生性腎小球腎炎由於MPGN常在上呼吸道感染之後急性起病,表現為急性腎炎綜合徵,甚至半數左右患者抗“O”鏈球菌感染的證據呈陽性,故應與鏈球菌感染後腎小球腎炎相鑑別後者常有肉眼血尿,而血補體水平在2個月內常恢復正常。本病肉眼血尿在發病後第1年內較為少見,而持續的低補體血症則應懷疑本病。另外鏈球菌感染後腎炎的病理常表現為毛細血管內增生性腎小球腎炎結合病理檢查不難區分。
Ⅳ型系統性紅斑狼瘡活動期,補體,尤其C3常降低,病理檢查有時也有系膜結構向基底膜和內皮間長入形成間位病變累及廣泛免疫複合物可以沉積於腎小球的各個部位,與本病Ⅰ型有些混淆,但是注意結合臨床其他表現和免疫螢光檢查的C1q的陽性程度以及血清免疫學檢查可加以鑑別。
Ⅰ型的病理檢查系膜區明顯擴張,可表現為結節性硬化灶,與糖尿病腎小球硬化或者輕鏈沉積病的光鏡改變相似,但是免疫螢光和電鏡的結果可以容易地將本病和其他疾病區分開,當然結合臨床表現,血生化和血清免疫學檢查就更容易鑑別。
應與其他的繼發性系膜毛細血管性腎炎相鑑別。如B肝相關性腎炎,根據病毒血清學及腎臟組織B肝病毒抗原標誌物可以鑑別。冷球蛋白血症臨床與病理均與該病相似,但很少見,並且前者有相應的全身表現病理有腎臟小血管炎和透明血栓形成提示為繼發性的病變。
鑑別診斷
 膜增生性腎小球腎炎
膜增生性腎小球腎炎1.糖尿病腎病 MPGN的結節狀損害出現在大多數腎小球中,而糖尿病腎病發生結節狀損害的小球相對較少,另外從免疫病理學上可以進行鑑別。
2.澱粉樣變腎病 HE剛果紅染色及電鏡下完全可以鑑別。
3.輕鏈腎炎 光鏡下與MPGN難以鑑別,免疫病理學可以明確區分。
4.狼瘡性腎炎 慢性低補體血症應與狼瘡性腎炎進行鑑別狼瘡性腎炎可以出現多種類型的病理學改變,如可出現類似於ⅠⅢ型MPGN樣的改變但狼瘡性腎炎在腎小球內可有IgGIgM、IgA、C3、C4C1q的沉積,即“滿堂亮”表現,而MPGN同時出現多種免疫球蛋白及補體沉積的情形罕見。
5.過敏性紫癜腎炎 可出現類似於MPGN樣的病理變化,鑑別的要點為紫癜性腎炎腎小球系膜區及毛細血管襻上有大量的IgA沉積,還可表現出皮膚紫癜、關節痛和腹痛等。
6.感染後腎炎 感染後腎炎與MPGN的Ⅰ型有時難以鑑別,但一般感染後腎炎的病程比較短。偶爾感染後腎炎也可發展為MPGN。
治療
 表3
表3Ⅰ型的治療,除糖皮質激素外,還可用其他藥物如免疫抑制藥和抗凝劑。
對於各年齡段MPGN患者,如腎功能正常且僅表現為無症狀輕度蛋白尿時,無須接受激素免疫抑制藥治療僅需每3~4個月隨訪1次密切觀察腎功能、蛋白尿和血壓控制情況。成人和兒童原發性MPGN患者,在尿蛋白>3g/d,腎功能損害及活檢發現腎間質病變時,方可給予激素、免疫抑制藥治療。
對於有蛋白尿(>3g/d)或腎功能損害的兒童原發性MPGN患者,大劑量糖皮質激素隔天40mg/m2,治療6~12個月後可能有效。如果無效則停止服用糖皮質激素建議密切隨訪,著重保守治療(即控制血壓、套用降低尿蛋白藥物和糾正代謝紊亂)。
對於有蛋白尿(>3g/d)或腎功能損害的成人原發性MPGN患者應給予阿司匹林(325mg/d)、雙嘧達莫(潘生丁)治療(75~100mg,2次/d),或二者聯合套用12個月,該治療方案如果無效則停用。重視能夠延緩腎功能衰退的因素和密切隨訪應是治療計畫的一部分。
幾項治療研究報導了隔天或每天口服糖皮質激素、靜脈注射大劑量糖皮質激素、及兩者聯合套用的結果:其中1項較嚴謹的研究證實:對兒童MPGN激素治療在減緩腎小球濾過率(GFR)下降和穩定腎功能方面有效80例患兒大部分為Ⅰ型MPGN,套用潑尼松(強的松)40mg/m2隔天口服,平均治療時間為13個月,61%的治療組患兒在研究階段腎功能穩定,而安慰劑組僅為12%。證明了兒童MPGN患者套用糖皮質激素有效。研究顯示兒童的MPGN早期套用大劑量潑尼松(強的松)可有效縮短病程。但尚有爭議,仍須用嚴格的隨機對照試驗來驗證。
Danadio等分別研究雙嘧達莫(潘生丁)、阿司匹林和華法林在MPGN治療中對腎臟轉歸(包括尿蛋白排泄率)和血小板半衰期(出血傾向)的影響。結果是尿蛋白排泄率降低,但GFR無明顯變化。Cattran等研究59例MPGN患者服用環磷醯胺華法林和雙嘧達莫(潘生丁)的療效治療時間18個月作者詳細說明了該項研究僅對Ⅰ型MPGN患者有較清晰的治療,而對Ⅱ型疾病沒有足夠有力的證據得出結論。
 飲食
飲食其他治療包括降脂、ACEI、ARB、低分子肝素等,近年有學者報導用霉酚酸脂(MMF)治療本病,顯示初步效果,但病例數尚少,且缺乏對照和長期觀察研究。
另外細胞毒藥物套用、血漿置換方法、中藥治療在一些研究治療中獲得一些療效。
臨床醫生在決定什麼類型的患者何時進行治療時必須考慮疾病的預期病程和結局,以及治療的利弊,腎功能不全的進行性發展和藥物治療引起的依從性差等因素。潑尼松隔天治療本病的情況見表3。
預後
 膜增生性腎小球腎炎
膜增生性腎小球腎炎總之,本病進入終末期腎病的個體差異比較大,Ⅰ型患者通常1/3可以自發緩解,1/3呈進行性發展還有1/3疾病遷延進展緩慢,但一直不能完全緩解。
文獻資料中提示原發性Ⅰ型病變的預後不良因素有:高血壓,腎功能損害,腎病綜合徵範圍的蛋白尿形成,腎活檢時發現細胞性新月體,合併動脈病變,有腎小管及間質的損害英國的Cameron發現Ⅰ型有腎病綜合徵範圍的蛋白尿者10年的生存率為40%,而非腎綜範圍蛋白尿患者的10年生存率為85%;但另有研究認為二者預後沒有差別,血尿甚至肉眼血尿對預後都無影響,年齡和性別也都不影響本病預後。
Ⅱ型較Ⅰ型的預後差這可能是由於Ⅱ型為緻密物沉積疾病,腎活檢常會發現新月體和小管間質病變。Ⅱ型很少發生臨床緩解兒童患者的臨床緩解率不足5%。患者通常在病程的第8~12年進入腎功能衰竭Ⅱ型患者在做腎移植以後常常會復發,尤其是腎移植前活檢就發現有新月體改變的患者。Ⅰ型在腎移植後也可能出現再發但是沒有Ⅱ型頻繁。
預防
本病3型病程轉歸基本相同預防要從自身健康著手,平時避免勞累,合理飲食科學鍛鍊,增強體質,提高機體免疫力,以防疾病發生。對於已患和出現併發症的病人,應對原發病及併發症進行積極有效的預防和治療。一旦發現感染,應及時選用對致病菌敏感強效且無腎毒性的抗生素治療,有明確感染灶者應儘快去除,以防腎功能不全的進行性發展。
