病因
 睪酮常規檢查
睪酮常規檢查一些家系分析的結果,發現一個嗅覺缺失的父親生育了嗅覺缺失和(或)性腺功能減退的兒子而所生的女兒性腺發育和嗅覺正常更為有趣的例子是父親是卡爾曼綜合徵患者,經過長期人絨毛膜促性腺激素治療後,結婚並生育了患卡爾曼綜合徵的兒子。這些家系例證與常染色體顯性遺傳一致。另一些家系則是祖代和父代家庭成員沒有發現異常,第3代的兒女中男性和女性都有嗅覺缺失和性腺功能減退患者,這種遺傳方式顯然符合常染色體隱性遺傳。此外,還有一些家系父親正常母親是攜帶者,生育的子女中,只有男性出現性腺功能減退
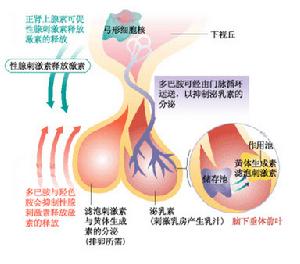 促性腺激素結構圖
促性腺激素結構圖和(或)嗅覺缺失而女兒結婚後,生育的女性子代表現正常,而男性子代是卡爾曼綜合徵患者,屬於X-連鎖遺傳這種遺傳的不均一性不僅表現在遺傳方式上,即使是同一遺傳方式也存在表達的不均一性即同一家系發病的成員中,可有單純性腺功能減退而無嗅覺缺失,或只有嗅覺缺失而無性腺功能減退;嗅覺缺失的程度也存在差異,一些受累家庭成員的嗅覺缺失是不完全的只有嗅覺減退。一個更為突出的例證是一對20歲同卵孿生兄弟,其中一個是典型的卡爾曼綜合徵患者,而另1個只有嗅覺缺失,生殖器官發育正常,血漿促性腺激素和睪酮水平正常。
發病機制
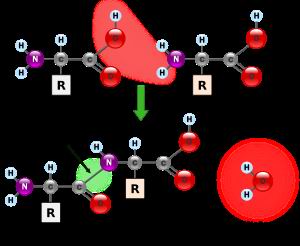 胺基酸結構圖
胺基酸結構圖鄰近基因的連帶缺失可引起卡爾曼綜合徵X-連鎖的魚鱗癬(STS基因缺失)、智慧型減退和(或)點狀軟骨發育不全KAL1基因的不同突變方式轉錄出不同的基因產物,後者與臨床表現的不均一性有關。現在已可套用Southern印跡技術分析胎兒的DNA在產前診斷X-連鎖型卡爾曼綜合徵。KAL1基因長約1.5Mb,編碼1個680胺基酸的糖蛋白,在功能上這個蛋白具有細胞外神經黏附分子的特性,可能是GnRH神經元從胎兒時期的嗅板遷徙到下丘腦內側底部的引路蛋白關於基因治療尚無可行的方案,但是KAL1基因及其編碼蛋白的結構已經闡明,有朝一日通過基因治療補充正常結構蛋白以預防卡爾曼綜合徵不是完全不可能的事情。至於常染色體顯性遺傳和隱性遺傳2種類型的致病基因現在仍所知甚少,是否在某條常染色體中存在著和KAL1相似的基因,還是KAL1基因亦與常染色體遺傳類型有關?此外,單純表達低促性腺激素性性腺功能減退而無嗅覺減退的患者是否亦是KAL1基因起著關鍵的作用?這些問題還有待於進一步的研究結果來回答。
臨床表現
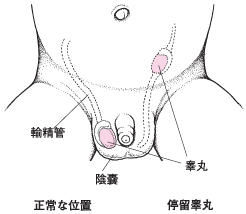 隱睪示意圖
隱睪示意圖診斷
 血清促性腺激素
血清促性腺激素鑑別診斷:
主要是特發性青春期延遲。特發性青春期延遲患兒生長遲緩,骨齡落後於實際年齡性幼稚血清促性腺激素和性激素水平低,以及促性腺激素對GnRH興奮無反應或反應減低等特點,與IHH非常相似,難以相互鑑別。多年來不少學者作了大量的研究,試圖尋找一種鑑別診斷試驗有效地將這2種情況鑑別開來。
檢查
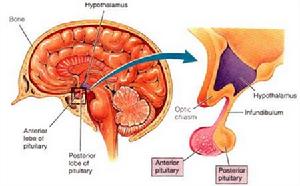 下丘腦結構圖
下丘腦結構圖2.GnRH興奮試驗無論是男性或女性患者,LH的分泌反應一般是減低的,少數患者完全無反應或反應正常。同一患者的LH反應可以和FSH反應不一致。
3.血清PRL基礎水平正常,PRL對促甲狀腺激素釋放激素(TRH)和氯丙嗪興奮試驗的反應一般正常,少數反應減低,個別反應過強。
4.患者的甲狀腺功能(臨床表現和TT4,TT3,FT4FT3和TSH)正常,TRH興奮TSH試驗一般反應正常,ACTH和皮質醇的晝夜節律正常,皮質醇對ACTH興奮的反應正常。
5.尿濃縮功能正常。
以上資料說明除下丘腦-垂體-性腺軸系外,腺垂體的PRL,GH,ACTH和TSH功能正常,神經垂體功能也正常。
預後
1.長期靜脈給藥治療會給患者生活帶來不便降低了患者對治療的順應性有發生靜脈炎的危險。
2.雌激素替代治療已有報導,長期大劑量服用雌激素使乳腺癌的發病率增高。子宮內膜癌的發病率增加4倍,在停藥後10年內仍有這種危險。
3.心肌梗死、肺梗死和血栓性靜脈炎的發病率增加。此外可有血壓增高、體重增加、水腫、高血鈣糖耐量減低和加重卟啉病等。
預防:
1.在雌激素替代治療期間應定期做乳腺婦科,心血管系統等方面的檢查。
2.性激素替代治療劑量要從小量開始,以避免骨骺過早閉合導致矮身材。
