流行病學
沃納(Werner)(1904)最早報導本病。兩性發病,男女之比為1∶1。雖然多數病例為家族性發病但也有散在發病者。所有病例出生時均正常,至幼兒期發育也都正常唯至學齡期或青春期生長突然停滯。發病與人種差異無關。多見於有血緣婚姻的子代,尤以堂兄妹間結婚者的子代居多。病因
本病系常染色體隱性遺傳性疾病,多見於有血緣婚姻的子代,尤以堂兄妹間結婚者的子代居多。本病基因定位於8p12~p11。發病機制
 沃納綜合徵
沃納綜合徵基於本病多有性功能減退和性腺發育不良,故推測可能與垂體功能低下有關。有人發現病人血鈣異常升高,皮膚和皮下組織多有鈣鹽沉著,因而構想本病發病可能與甲狀旁腺功能亢進有關。還有人認為和肝臟滅活氫化可的松的功能不佳有關,因而出現可的松過剩和抗蛋白同化作用增強,呈現為生長停滯、組織萎縮和糖尿病。但以上構想均未獲得廣泛的承認。
本病的主要病理改變為:鈣化性主動脈粥樣硬化,常伴以周圍動脈粥樣硬化皮膚和皮下軟組織明顯萎縮。全身性細長體型和睪丸嚴重萎縮。惡性腫瘤發生率高。
根據Fleischner描述本病的表皮角化過度,皮膚附屬器變性,有不同程度的真皮纖維化和透明變性,管腔狹窄腎上腺球狀帶傾向於增寬,全身各組織均可發生纖維化和其他結締組織改變。 由於肌纖維破壞喪失和纖維化而引起肌肉萎縮,是本病的常有組織學改變此外尚有肌纖維腫脹、橫紋消失、肌纖維的大小和形狀不規則等。
主動脈和大血管鈣化是本病常見的病理改變,所有病例均有與年齡不成比例的嚴重動脈粥樣硬化常波及冠狀動脈而發生心肌梗死。也可見有動脈中層鈣化和透明性變動脈硬化。心血管病變最明顯的特徵為主動脈或二尖瓣嚴重鈣化。部分病例可發生垂體嫌色細胞瘤,也有的發生垂體嗜鹼性腺瘤。
在腦組織中可見有因動脈硬化所致的腦皮質萎縮但無明顯脂褐質色素沉著和澱粉樣小體周圍神經無異常發現。
臨床表現
兩性發病,男女之比為1∶1。雖然多數病例為家族性發病,但也有散在發病者。所有病例出生時均正常,至幼兒期發育也都正常,唯至學齡期或青春期生長突然停滯。四肢和軀幹同時發育停滯,故可保持均勻對稱、身材矮小體型。鼻樑高聳,呈特有的鳥嘴樣尖鼻(鷹鉤鼻)。
1.皮膚損害 毛髮變白為本病最早出現的體徵,在10~20歲時就出現,通常從頭皮和眉毛開始,並有進行性脫髮,眉毛、陰毛亦脫落至40歲時全部頭髮均可變白或成為禿頭。顏面四肢皮膚萎縮,呈老年人樣面容。至青春期,四肢皮膚、皮下組織和肌肉可發生向心性瀰漫性萎縮,因此,皮膚拉緊呈過度伸展樣外觀,並與皮下組織緊密結合在一起。這種改變上肢多於下肢但軀幹改變不甚明顯。局限性角質增生亦為本病的常見皮損多發生於手掌、足底部有時可因發生胼胝(雞眼)而引起局部疼痛。在足外側踝部及跟腱部等易受壓迫之處可形成潰瘍而且不易治癒。其他皮損尚有毛細血管擴張皮膚色素脫失性萎縮和全身性軟組織鈣化後者通常為血管周圍鈣化。
2.骨關節病變 由於四肢皮膚萎縮、拉緊皮下組織纖維化及局部血管供血障礙,結果可致受累關節運動受限肢端萎縮及強直變形。另外,本病的特徵性異常表現為全身 性骨質疏鬆。由於全身性發育過早停滯,故常出現手腳過小、四肢短小並伴有肌肉組織消瘦等。
3.心血管病變 常為全身性主要特徵為嚴重的心血管病變,表現為局部供血不全症狀如冠心病等。
4.內分泌異常 本病偶有內分泌功能紊亂表現,例如伴有糖尿病視網膜病糖尿病的發生,又可加重血管病變偶爾還可發生糖尿病昏迷。本病還常發生性功能低下,男性表現為性器官發育不全、性慾低下,女性表現為月經過早來潮,月經過少,過早閉經,大小陰唇、陰道、內生殖器及乳房發育不良或不全。
5.神經系統異常 1/3的病人有輕度神經系統症狀,其中最重要的表現是累及肢體遠端的肌病型肌萎縮,遠端深部腱反射消失,部分病例可出現感覺異常。約半數病人有智力低下,可伴有幼兒型情緒;少數病例可有精神症狀和癲癇大發作本病並發非癌腫性腫瘤的發生率較高,其中最常見的為腦膜瘤和神經鞘肉瘤。
6.五官病變 近半數病人有異常高調的說話聲音。喉鏡檢查可見聲帶上或其附近血管有萎縮性、擴張性或隆起的淺表性改變,聲帶有黏膜充血區,這種黏膜改變即可造成高調聲音。
白內障是本病的主要特徵,多發生在20~30歲,故稱青年白內障。晶體渾濁呈星芒狀,常為雙側性,多先出現於晶狀體的後極。
併發症:
本病除容易並發腦瘤外,並發肝癌乳房腫瘤、纖維肉瘤、黑色素瘤的發生率也很高。個別病例可發生肝大,並伴有功能異常和貧血等症狀。
診斷
當病情處於慢性進行階段,臨床症狀和體徵已較典型時,診斷並不困難。但在早期診斷可能比較困難,可根據毛髮過早變白、特殊面容和特徵性體型等臨床特徵,結合實驗室和X線發現進行診斷。Irwin將本病的臨床表現歸納為以下4組:
1.特徵性體型和體質 ①青春期出現矮小體型;②軀幹短胖和四肢纖細;③有鳥嘴樣尖鼻。
2.過早衰老 ①毛髮過早變白;②過早禿髮;③聲音變尖而弱;④動脈粥樣硬化;⑤皮膚萎縮;⑥青年白內障。
3.硬皮病樣變化 ①皮膚和皮下組織萎縮;②局部角化症;③足背皮膚過緊;④足踝部跟腱、足跟和趾潰瘍。
4.其他表現 ①糖尿病傾向;②性腺發育不良;③骨質疏鬆;④局部鈣化。
5.同胞間偶有發病傾向 在臨床上,可以根據這四組特徵做出診斷。但必須指出,第2項中的③和第3項中的④及第4項中的①,並不是必備的診斷條件。另外,上肢肢端也可發生第4項中的③與④。
鑑別診斷:
診斷應注意本綜合徵與肌緊張營養不良的區別,後者肌肉萎縮以顳肌最為明顯,病人呈“斧頭”面容(Hatchet-face),嚴重時下頜肌和頸部肌群可嚴重萎縮肌緊張反應是二者的最重要鑑別點。還應注意本綜合徵與硬皮病、指端硬化症及伴掌皮膚硬化的肢端硬化症相鑑別。根據這些疾病有無毛髮變白、過早禿髮以及有無沃納綜合徵的特徵性面容與體型,比較容易鑑別。
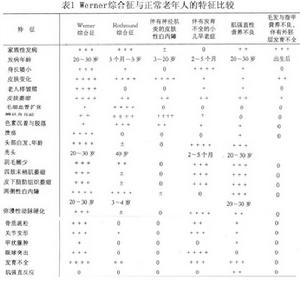
沃納綜合徵必須與自然老化仔細鑑別(表1)。另外,還必須與盧斯曼(Rothmund)綜合徵,伴有神經肌炎的皮膚性白內障(Andogsky綜合徵),伴有發育不全的小兒早老症(Hutchinson綜合徵),毛髮、指甲營養不良伴有外胚層發育不全(Ellis-van Greveld綜合徵)肌強直營養不良(Steinert綜合徵)等,加以區別。
檢查
實驗室檢查:
1.生物化學檢查 多數病人血脂分析示膽固醇、β-脂蛋白和三酯甘油升高。尿肌酐、胺基酸升高。
2.免疫學檢查 T細胞減少及T細胞免疫功能低下抗淋巴細胞抗體可為陽性。
其它輔助檢查:
1.X線檢查:脊椎及四肢骨質疏鬆,在肢體軟組織特別是在骨性突起周圍,有線狀與圓形的鈣化陰影。在主動脈、主動脈瓣二尖瓣和冠狀動脈,亦可有廣泛的鈣化,並有心臟增大與充血性心力衰竭的徵象。
2.皮損活檢化學分析示羥脯氨酸、氨基葡萄糖升高。
治療:
目前無特效療法,只能對症治療併發症,尤其應及時長期對症治療動脈硬化、冠心病可套用擴血管藥物加降血脂製劑,以延緩病變的發展。可用蛋白同化激素促進或維持第二性徵,抑制骨質疏鬆的發展速度及減輕萎縮。據報導,用EDTA降低血鈣可減輕軟組織鈣化。對於白內障,必須特別謹慎地施行手術治療,以免引起角膜變性、繼發性青光眼和完全失明。由本綜合徵所引起的糖尿病有抗胰島素傾向,適當控制飲食和口服降糖藥物通常足以控制血糖。一旦確診為本綜合徵,就應仔細檢查是否合併腫瘤,以便及時手術切除
預後預防
預後:
預後較好,存活年齡在31~63歲不等。主要死亡原因為腦血管意外腫瘤、糖尿病昏迷、肝功能衰竭和貧血。
預防:
1.努力降低人群中遺傳病發生率,必須採取有效的預防措施,針對個體,採取通常的措施包括婚前檢查遺傳諮詢、產前檢查和遺傳病的早期治療。
2.保證男女雙方婚後生活幸福、後代健康的重要環節是不要近親婚配。降低人群遺傳病的發生率,降低有害基因的頻率,及減少傳遞機會。
