
病因
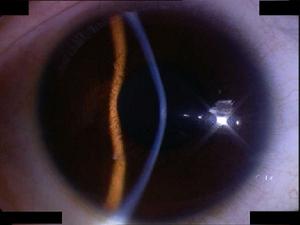 圓錐角膜
圓錐角膜發病機制
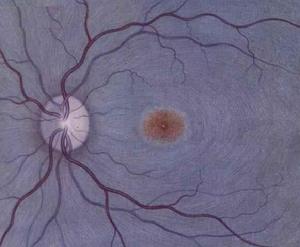 角膜上皮細胞
角膜上皮細胞類似ICE綜合徵的膜理論曾被認為是PPD繼發性閉角型青光眼的發病機制。異常內皮細胞或上皮細胞及其基底膜樣物質從周邊角膜向下越過虹膜角膜角和延伸到虹膜表面,隨後此膜收縮導致虹膜角膜粘連房角關閉、瞳孔移位、葡萄膜外翻及虹膜萎縮。繼發開角型青光眼的機制尚未肯定。虹膜角膜角鏡檢查及超微結構研究發現這些患者的虹膜恰好在鞏膜嵴之前嵌入後部小梁網,推測虹膜起著壓迫作用,導致小梁網間隙及小樑柱萎陷和增加房水流出阻力。這些發現提示虹膜角膜角存在發育異常和類似某些先天性青光眼的發病機制。
臨床表現
 眼角膜
眼角膜2.眼部表現 主要為角膜病變,虹膜和虹膜角膜角也可受累。裂隙燈檢查所見PPMD的形態改變包括內皮小的聚集性小泡較大地圖狀水泡樣改變,後彈力層呈灰色混濁或大小不等的斑塊混濁而無點狀角膜變性。這些改變位於深部角膜後彈力層水平。
本病最單純的形式為聚集性小泡,又稱為後部皰疹或線狀小泡。2~20個小的(0.2~0.4mm)不相連的圓形病變聚集一處,在光線照射下形似成堆的小泡或水皰,圍以瀰漫的灰色暈。此病變可以出現在角膜後的任何地方,可長期保持穩定,亦可增多或退行,對視力則無影響。較重時,小泡聚集成大的地圖樣病變。較大的地圖樣病變為聚集性小泡的較嚴重型,其灰色暈較濃且有時呈結節狀,其圓形或橢圓形小泡間分界則較模糊。病變分布可多種多樣,自周邊部環狀至局部楔形,直至角膜後面瀰漫的硬幹酪狀圖案。在寬光帶照明下,可見病變有2個半透明的、邊緣呈扇形花樣突起的嵴組成。用後照明法可見整個角膜後部呈金屬箔光澤的橘皮狀。基質及上皮水腫與其他類型的角膜水腫相似。基質水腫自後方開始,逐漸變濃,當上皮表面變為不規則時,視力下降。有些病例,寬的周邊部虹膜角膜粘連占據角膜後面周邊部1mm範圍,有時伴有玻璃體膜,自粘連部向下伸展至虹膜表面,造成虹膜上皮外翻及瞳孔變形,並可見虹膜萎縮。裂隙燈檢查初始角膜仍透明,以後出現多形性地圖樣不透明體聚集,增厚的後彈力層如後帶狀伴贅生物,導致內皮失代償而引起角膜水腫。少數出現虹膜周邊前粘連,引起瞳孔變形、色素層外翻及繼發性青光眼。
3.全身症狀 典型的PPMD是伴有全身異常。已有報導PPMD可能是全身基底膜疾病的一種證據,並已發現與A1port綜合徵(是具有先天性腎炎和聽力喪失的基底膜疾病)有密切關係。患有Alport綜合徵的病人必須進行全面的眼部檢查,包括特殊顯微鏡檢查,而患有PPMD的病人也要進行腎功能評價和聽力檢查。
4.繼發青光眼 在PPMD病人中約13%的病人合併青光眼。青光眼臨床表現有以下幾種類型:成人開角型青光眼、成人閉角型青光眼及嬰幼兒型青光眼。具有開角型和嬰幼兒型青光眼的病人虹膜角膜角鏡檢查正常,具有高位虹膜房角關閉的青光眼虹膜角膜角鏡檢查可見虹膜角膜粘連,形成絲狀或柱狀粘連房角關閉的眼同時可以有虹膜萎縮和瞳孔移位,一旦出現沿房角60°~120°範圍內寬型粘連,通常出現眼壓升高。
併發症:帶狀角膜病變、角膜失代償、虹膜周邊前粘連等。
診斷:根據臨床表現並有遺傳學特徵,可以確定診斷,但需要與相似疾病進行鑑別。
鑑別診斷:PPMD常與角膜後營養不良的其他類型易相混淆,例如Fuch內皮營養不良、先天性遺傳角膜營養不良和後無定型的角膜營養不良。後者通過灰白擴散的後基質的片狀混濁,偶爾虹膜伸延到360°Schwalbe線以及各種虹膜異常,但是無青光眼。當角膜虹膜粘連出現,需同時考慮Axenfeld-Rieger綜合徵和ICE綜合徵,前二者中有許多較細的虹膜絲附於突出的Schwalbe環上。PPMD往往與先天性青光眼角膜後Haab’s線相混儘管後者可以通過邊緣的薄區域與之鑑別內皮細胞顯微結構在鑑別PPMD和其他有前角膜異常的疾病方面是有價值的。
檢查
1.遺傳學檢查 確定其遺傳方式。
2.病理學檢查 套用光鏡檢查可現後彈性膜紡錘狀贅瘤,並於Descemet膜水平有小泡形成。一部分PPMD的病例可形成較寬的周邊前粘連,向前進展附著於Schwalbe線或角膜上,並且可合併瞳孔異位、虹膜色素外翻及虹膜稀疏,尚有病例可見到半透明的玻璃膜從角膜後面擴展到虹膜上電鏡觀察顯示角膜內皮和深層Descemet膜異常。房角可被角膜內皮細胞覆蓋,但異位的角膜內皮細胞形態與Chandler綜合徵不同,具有上皮細胞特徵,包括微絨毛突、一些線粒體細胞質角質絲的存在和多層細胞間的橋連線。典型的表現為Descemet膜變厚並被多層膠原覆蓋,此外可有異常內皮細胞成纖維細胞或上皮細胞的變異細胞充填。角膜混濁或水腫的臨床區別取決於異常上皮細胞取代正常內皮細胞鑲嵌的程度,特殊顯微技術和細胞培養技術可判斷PPMD患者內皮細胞和上皮細胞的存在。另外這種膜及帶有上皮樣細胞的內皮亦見於虹膜上。
治療
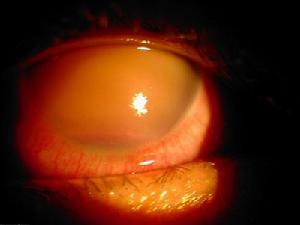 青光眼
青光眼PPMD和由於虹膜角膜粘連而繼發青光眼的病人實行角膜移植手術是困難的虹膜角膜粘連的眼最後都繼發青光眼,這對角膜移植能否成功是至關重要的影響因素。對於這類病人那些人只有到必需時才考慮行角膜移植術。
PPMD合併有青光眼的治療,方法類似ICE綜合徵的病人。可考慮用縮瞳藥打開房角,但實際效果並不肯定β阻滯藥和碳酸酐酶抑制藥及腎上腺素製劑在開角型和閉角型青光眼同樣有效。
雷射小梁成形術很少有效如果使用,小心由於膜的增生跨越房角加速房角關閉。套用最大劑量藥物治療,眼壓仍不能控制,則需進行濾過手術與其他類型慢性青光眼比較,手術成功率低。
併發症
帶狀角膜病變、角膜失代償、虹膜周邊前粘連等。
