主要內容
簡介希佩爾·林道綜合徵(Von Hippel–Lindau disease,VHL綜合徵),或譯為希佩爾·林道病,是一種罕見的常染色體顯性遺傳性疾病,表現為血管母細胞瘤累及小腦、脊髓、腎臟以及視網膜。其若干病變包括腎臟血管瘤、腎細胞癌以及嗜鉻細胞瘤等。疾病是因位於染色體3P25.3的VHL抑癌基因發生突變所致。
希佩爾·林道綜合徵還有其他不常見的命名,包括:視網膜血管瘤,囊腫性視網膜血管瘤(小腦及視網膜內血管瘤樣囊腫形成),家族性小腦-視網膜血管瘤,小腦視網膜血管母細胞瘤,視網膜小腦血管瘤。
表現及症狀
VHL的臨床表現和症狀包括血管瘤、血管母細胞瘤、嗜鉻細胞瘤、腎細胞癌,胰腺囊腫(胰腺漿液性囊腺瘤)以及咖啡牛奶斑。37.2%的VHL患者表現為血管瘤,且通常累及視網膜,由此產生的失明十分常見。其他臟器也會受累,由此帶來的中風、心臟病、心血管疾病也相當多見。
遺傳學觀點
希佩爾·林道綜合徵是因位於3號染色體短臂(3P25-26)的VHL抑癌基因突變所致。
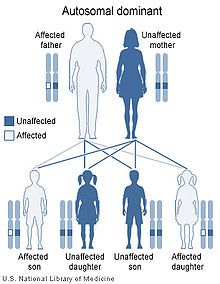 VHL綜合徵是常染色體顯性遺傳模式
VHL綜合徵是常染色體顯性遺傳模式遺傳性VHL基因突變占到了80%。而大約20%的病例,是由於基因在生殖細胞(卵子或精子)形成時或在胎兒生長早期發生新的突變所致,這種情況非常罕見,因為在一個細胞中,原先均正常的兩個等位基因同時發生突變的機率很低。無論是新的突變還是遺傳性突變,前面提及的第二次突變對於腫瘤的發生是不可缺的。
疾病的發病年齡、對器官的影響和病變程度在不同患者中差別很大。這提示了第二次突變可發生在不同類型的細胞中,且發生在人生的不同階段。治療
如今沒有方法可以逆轉患者VHL基因的突變。儘管如此,早期識別VHL綜合徵特徵性的臨床表現並進行治療,這能從本質上減少併發症的發生並提高患者的生活質量。
發現歷史及相關事件
發現歷史德國眼科醫生Eugen von Hippel在1904年第一個描述了眼球內血管瘤的病例。Arvid Lindau在1927年描述了小腦及脊髓內血管瘤的病例。疾病也因此用兩人的姓氏命名。
相關事件一些McCoy家族(即美國阿巴拉契亞和Hatfield家族有世仇的McCoy家族)以及Elliott家族的後裔患有VHL。在一篇美聯社的報導中,一位來自范德堡大學的內分泌學家推測,Hatfield家族和McCoy家族間潛在的敵意,有一部分可能是由於VHL疾病導致的。報導指出,McCoy家族有脾氣暴躁的傾向,因為家族中的許多人患有嗜鉻細胞瘤,產生過多的腎上腺素,從而導致暴躁的脾氣。但是,嗜鉻細胞瘤產生大量的腎上腺素,更多表現為驚恐發作而不是暴力襲擊。未經治療的患者會有罹患心血管疾病,心臟病以及中風的危險。只有大約20%的VHL患者有嗜鉻細胞瘤。
