概述
己糖激酶缺乏症(hexokinase deficiency, HKD)是常染色體隱性遺傳疾病。1967年,Valentine等最先報導了3例,現已報導12個無親緣關係家族中的20例患者 其中1例7歲男孩為中國血統。
流行病學
全球報導不足20個家系,分布於歐洲、地中海、斯堪的那維亞和東半球,國內有隻有1個家系報導。
病因
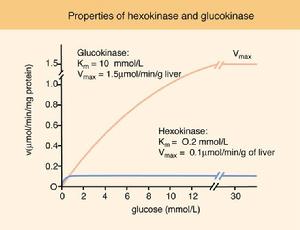 己糖激酶與葡萄糖酶比較
己糖激酶與葡萄糖酶比較發病機制:HK為一分子量108kD的單體,是葡萄糖無氧糖酵解途徑第1個催化酶 是該途徑關鍵性限速酶之一 HK有4種異構體(Ⅰ、Ⅱ Ⅲ和Ⅳ),紅細胞主要是Ⅰ型HK 它又可分為Ⅰa、Ⅰb和Ⅰc。HKⅠa主要存在於幼紅細胞中 成熟紅細胞中水平較低,是一種細胞年齡依賴性酶 HKⅠ型存在於淋巴細胞和血小板 HK缺乏症患者淋巴細胞HKⅠ型量減少 HKⅢ型代償性增高,血小板僅只有正常HK活性的20%~35%。凝血酶刺激後,儘管糖酵解受抑制和高密度顆粒分泌受損 但血小板代謝的缺陷無臨床意義 。
HK Ⅰ型結構基因定位於10p11.2。最近已從一個成人腎DNA文庫中分離出了編碼人HK的cDNA克隆 HK缺乏症的遺傳方式為常染色體隱性遺傳 HK缺乏症時,葡萄糖代謝中間產物和ATP生成減少 2,3-DPG可能減少或正常 。
症狀
 己糖激酶缺乏症
己糖激酶缺乏症併發症:目前暫無相關資料。
診斷:己糖激酶缺乏症的確認有賴於紅細胞HK活性分析 。
鑑別診斷:目前暫無相關資料。
檢查
 己糖激酶缺乏症
己糖激酶缺乏症2.HK活性測定,血小板和白細胞HK活性降低。
3.紅細胞滲透脆性增高,自身溶血試驗正常或加葡萄糖和ATP部分糾正後而致增高(Ⅰ型) 。
其它輔助檢查:目前暫無相關資料。
相關檢查:2,3二磷酸甘油酸胰構酯;紅細胞三磷酸腺苷;紅細胞己糖激酶;自身溶血試驗; 葡萄糖;血小板 。
治療
預防
預後:預後一般較差 。
預防:應注意預防提倡優生,進行婚前和產前檢查。
