
合成步驟
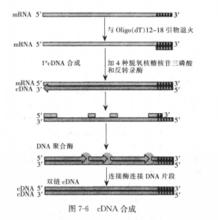
1.cDNA第一鏈的合成 所有合成cDNA第一鏈的方法都要用依賴於RNA的DNA聚合酶(反轉錄酶)來催化反應。商品化反轉錄酶有從禽類成髓細胞瘤病毒純化到的禽類成髓細胞病毒(AMV)逆轉錄酶和從表達克隆化的Moloney鼠白血病病毒反轉錄酶基因的大腸桿菌中分離到的鼠白血病病毒(MLV)反轉錄酶。AMV反轉錄酶包括2個具有若干種酶活性的多肽亞基,這些活性包括依賴於RNA的DNA合成,依賴於DNA的DNA合成以及對DNA-RNA雜交體的RNA部分進行內切降解(RNA酶H活性)。MLV反轉錄酶只有單個多肽亞基,兼備依賴於RNA和依賴於DNA的DNA合成活性,但降解DNA—RNA雜交體中的RNA的能力較弱。且其熱穩定性較AMV反轉錄酶差。MLV反轉錄酶能合成較長的eDNA(如大於2~3kb)。AMV反轉錄酶和MI。V反轉錄酶利用RNA模板合成cDNA時的最適pH、最適鹽濃度和最適溫度各不相同.所以合成第一鏈時相應調整條件是非常重要的。
AMV反轉錄酶和MLV反轉錄酶都必須有引物來起始DNA的合成。cDNA合成最常用的引物是與真核細胞mRNA分子3’端poly(A)結合的12~18個核苷酸長的oligo(dT)。
2.cDNA第二鏈的合成 cDNA第二鏈的合成方法有以下幾種:
(1)自身引導法。合成的單鏈eDNA3’端能夠形成一短的髮夾結構,這就為第二鏈的合成提供了現成的引物。當第一鏈合成反應產物的DNA—RNA雜交鏈變性後利用大腸桿菌DNA聚合酶I Klenow片段或反轉錄酶合成eDNA第二鏈,最後用對單鏈特異性的S1核酸酶消化該環,即可進一步克隆。但自身引導合成法較難控制反應,而且用S1核酸酶切割髮夾結構時無一例外地將導致對應於mRNA5’端序列出現缺失和重排,因而該方法很少使用。
 cDNA合成
cDNA合成(2)置換合成法。該方法利用第一鏈在反轉錄酶作用下產生的DNA RNA雜交鏈不用鹼變性,而是在dNTP存在下。利用RNA酶H在雜交鏈的mRNA鏈上造成切口和缺口。
從而產生一系列RNA引物,使之成為合成第二鏈的引物。在大腸桿菌DNA聚合酶工的作用下合成第二鏈。該反應有3個主要優點:①非常有效;②直接利用第一鏈反應產物,無須進一步處理和純化;③不必使用S1核酸酶來切割雙鏈cDNA中的單鏈髮夾環。
3.cDNA合成技術 以Riboclone M-MLV CDNA合成技術為例。
Riboclone M—MLV cDNA合成系統採用M—MLV反轉錄酶的RNase H缺失突變株取代AMV反轉錄酶,使合成的cDNA更長。該系統的第一鏈合成使用M-MLV反轉錄酶,cDNA第二鏈合成採用置換合成法,採用RNaseH和DNA聚合酶I進行置換合成,最後用T4 DNA聚合酶切去單鏈末端,方法簡便易行。其基本步驟為先合成第一鏈:
(1)取一滅菌的無RNA酶的eppendorf管,加入RNA模板和適當引物,每RNA使用0.5ug引物(如使用Not I引物接頭,用0.3ug),用HO調整體積至15ul,70℃處理5min冷卻至室溫,離心使溶液集中在管底,再依次加入:
5X第一鏈緩衝液5ul。
RNasinRNA酶抑制劑25U
M—MLV(H)反轉錄酶200U
H2O調至總體積25ul
(2)用手指輕彈管壁,吸取5ul至另一eppendorf管,加入2~5ulCi[α一 P]dCTPdCTP(>400Ci/mmol),用以第一鏈同位素摻入放射性活性測定。
(3)37℃(隨機引物)或42℃(其他引物)保溫1h。
(4)取出置於冰上。
(5)摻入測定的eppendorf管加入95ul 50mM EDTA終止反應,並使總體積為100ul。可取90ul進行電泳分析(先用苯酚抽提)。另l0ul進行同位素摻入放射性活性測定。
(6)第一鏈合成eppendorf管可直接用於第二鏈合成。
以上25ul反應總體積中所用RNA量為1ug,如合成5ugRNA,則可按比例擴大反應體積。倒5ugRNA使用125uL總體積進行合成。
第一鏈合成後再進行第二鏈合成:
(1)取第一鏈反應液20uL,再依次加入:
10X第二鏈緩衝液20uL
DNA聚合酶123ul。
RNaseH0.8ul
H2O加至終體積為100/uL
(2)輕輕混勻,如需進行第二鏈同位素摻入放射性活性測定,可取出10uL至另一eppendorf管,加入2~5uLCi[α一 P]dCTP。
(3)14℃溫浴2h(如需合成長於3kb的cDNA,則需延長至3~4h)。
(4)摻入測定eppendorf管中加入90ul 50mM EDTA,取10ul進行同位素摻入放射性測定.餘下的可進行電泳分析。
(5)將cDNA第二鏈合成的反應液70℃處理10min,低速離心後置於冰上。
(6)加入2uL T4 DNA聚合酶,37℃溫育10min。
(7)加入10uL 200mmol/L EDTA終止反應。
(8)用等體積苯酚:氯仿抽提cDNA反應液,離心2min。
(9)水相移至另一eppendorf管,加入0.5倍體積的7.5mol/l醋酸銨(或0.1倍體積的1.5mol/L醋酸鈉,pH5.2),混勻後再加入2.5倍體積的冰冷乙醇(一20℃),一20℃放置30min後離心5min。
(10)小心丟去上清液,加入0.5mL冰冷的70%乙醇,離心2min。
(11)小心移去上清液,乾燥沉澱。
(12)沉澱溶於10~20ul TE緩衝液。
分子克隆
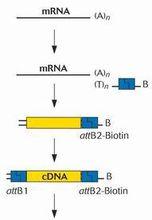 cDNA
cDNA已經製備好的雙鏈cDNA和一般DNA一樣,可以插入到質粒或噬菌體中。為此,首先必須連線上接頭(linker),接頭可以是限制性內切酶識別位點片段,也可以利用末端轉移酶在載體和雙鏈cDNA的末端接上一段寡聚dG和dC或dT和dA尾巴,退火後形成重組質粒,並轉化到宿主菌中進行擴增。合成的cDNA也可以經PCR擴增後再克隆入適當載體。
