基本內容
高血氨症 肝功能嚴重損壞時尿素合成障礙導致血氨濃度升高
【病因和發病機制】
本病代謝缺陷是尿素循環中有關的酶活性缺乏。其中任何一種酶活性的完全或部分缺乏,都會導致其底物在體內蓄積,以致血氨含量明顯升高。
【病理】
嚴重的高血氨症者有明顯的腦萎縮,皮質變薄而透明,側腦室和第3腦室擴大。鏡檢下見軟腦膜膠原增厚,皮質細胞有多處壞死和囊性變,並見有特異的原漿型星形細胞增大及增多,細胞核形大而透明,有細胞內顆粒,與AlzheimerⅡ型膠質細胞類似。除了腦組織以外,肝臟和其他內臟一般沒有任何組織病理學改變,但各型之間稍有不同。
【臨床表現】
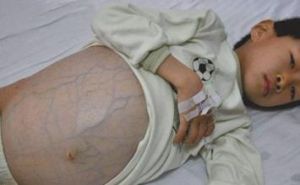 高血氨症
高血氨症1.氨甲醯磷酸酯合成酶(carbamyI phosphatesynthetase,CPS)缺乏症(高血氨症I型) 由Free-man等(1964)首先描述。患兒常從新生兒期出現嘔吐、嗜睡、全身肌張力低、抽搐、脫水及酸中毒等。餵飼蛋白類食物後則症狀明顯加重。常迅速死於酮症酸中毒。。肝組織也可有廣泛性脂肪浸潤。本病的病因已證實有CPS酶活性缺乏,尿素循環的其他酶類活性正常。近年來,又有人發現另一種症狀相類似的高血氨症,但其代謝缺陷起因於N-乙醯谷醯胺合成酶的缺乏,稱之為I。型。
2.鳥氨酸氨甲基轉移酶(ornithine transcar-bamylase,OTC)缺乏症(高血氨症Ⅱ型) 由Rus-sell等(1962)首先報告。本病僅見於男孩,符合X連鎖隱性遺傳。臨床表現與第1型相似,但起病較晚。病理檢查見腦組織中有大量AlzheimerⅡ型膠質細胞增生。實驗室檢查除了有血氨增高外,腦脊液中氨含量也有明顯增高。肝臟中除了OTC酶活性缺乏以外,有時CPS酶活性也可同時降低。
3.瓜氨酸血症(citrullinemia) 由Mc Murray等(1963)發現。本病的代謝缺陷是肝細胞精醯琥珀酸合成酶缺乏,不能使瓜氨酸合成精醯琥珀酸所致,其突變基因在9q44。多從出生後不久即發生高血氨症狀,包括頻繁嘔吐、全身性抽搐、運動和精神發育遲滯等。大都在嬰兒期死亡,但輕症患兒也可存活多年,而且智慧型發育正常。實驗室檢查除高血氨外,血中瓜氨酸、同型瓜氨酸含量明顯增高,有時谷醯胺、丙氨酸和肌肽等也可相應增多。尿中也排出瓜氨酸等大量含氮物質。
4.精醯琥珀酸尿症(argininosuccinic aciduria) 由Allan等(1958)首先發現。代謝缺陷是缺乏精醯琥珀酸酶,從血漿透析物或皮膚成纖維細胞培養中可測定其活性,突變基因在7qCen-q11.12。特點是精神發育遲滯,並伴有抽搐,間歇性共濟失調,全身肌張力低下和肝腫大。病兒頭髮較脆,並且扭結成團,是本病的特徵之一,尿中排出大量的精醯琥珀酸,是診斷的主要根據。有人按其起病早晚和病情緩急,分為新生兒變異型(起病急,多在1周內死亡),亞急性型(起病於嬰兒期,病程較緩)和晚髮型(起於兒童期或少年期,有典型的頭髮和神經症狀)。
5.高鳥酸血症(hyperornithinemia) 由施益安(Shih)等(1969)發現。臨床表現有嬰兒期餵養困難,精神和運動發育遲滯,和第1型相似,但常有肌陣攣性癲癇發作,症狀時輕時重,有些患兒伴有出血傾向。血中除氨含量增高外,還可見鳥氨酸,賴氨酸增高。同型瓜氨酸也增多。尿中可見有大量同型瓜氨酸。肝功能方面,GOT活性增高,腦磷脂絮狀試驗(CCFT)多強陽性。從末梢血白細胞培養中,也可測出CPS酶活性缺乏,但還有鳥氨酸-I)-氨基轉移酶的缺乏,與第1型不同的是其突變基因OAT在10q26。近來還有人推測可能由於線粒體中鳥氨酸易位物質(translocator)的異常,尚待證實。
6.高精氨酸血症(argininemia) 由Terheggen等(1969)首先報告。代謝缺陷是精氨酸酶活性缺乏,不能裂解精氨酸為尿素並加入鳥氨酸代謝循環,突變基因在6qz。。臨床特點為嬰兒期抽搐,智慧型發育遲滯,伴發作性嘔吐。有些患兒出現痙攣性癱和手足徐動症等。血中精氨酸含量可高出正常兒(3歲以下<10tLmol/L)7~10倍,同時腦脊液和尿中精氨酸也增多,尿中肌酐排出量增高。
7.高賴氨酸血症(hyperlysinemia) 由Woody(1964)首先報導。代謝缺陷是賴氨酸a酮戊二酸還原酶活性缺乏。表現有精神和運動功能發育停滯,身材短小,伴全身肌張力減低及嬰兒期驚厥等。實驗室檢查可見血氨增高,同時血、尿和腦脊液中賴氨酸含量也明顯增高,而其他胺基酸含量正常。
8.二鹼基氨酸尿症(dibasic aminoaciduria) 由Whelan和Seriver(1968)首先報告。可能為常顯遺傳。目前認為本病是一種胺基酸轉運功能的廣泛缺陷所致。l}缶床表現有生長發育停滯,餵養困難,對蛋白質攝入不能耐受。血中除了氨含量增高外,還可見單鹼基胺基酸增多,賴、精、瓜等二鹼基胺基酸減少。尿中二鹼基胺基酸則大量排出。
