 脊髓性肌肉萎縮
脊髓性肌肉萎縮流行病學
SMAⅠ~Ⅲ型稱為兒童型SMA,其群體發病率為1/6000~1/10000。SMA-Ⅰ型約l/3 病例在宮內發病,其母親可注意到胎動變弱。半數在出生1個月內起病,幾乎所有病例均在5 個月內發病。發病率約為1/10000 出生嬰兒,男女發病相等。SMA-Ⅱ型發病較SMA-Ⅰ型稍遲,通常於1 歲內起病,極少於1~2 歲間起病。發病率與SMA-Ⅰ型相似。SMA-Ⅲ型一般於幼兒期至青春期起病,而多數於5 歲前起病。20~30 歲以上起病的SMA,歸為第Ⅳ型,其群體發病率約為0.32/10000。
併發症
不同類型SMA 的症狀體徵可以是本病表現,也可以看作本病併發症。另外,應注意繼發的肺部感染、尿路感染、褥瘡等。 預後及預防(查看內容)預後:本病多數預後不良。SMA-Ⅰ型預後最差,約95%死於出生後18 個月。SMA-Ⅱ型具有相對良性的病程,多數可活到兒童期,個別活到成年。SMA-Ⅲ型預後良好,尤其是女性患者。生存期通常能達到成年,許多患者能有正常壽命。表現較嚴重病例往往為男性患者。SMA-Ⅳ型預後相對良好,行走能力常可保持終生。慢性不對稱型SMA 自然病程較長,甚至超過30 年。病因病理
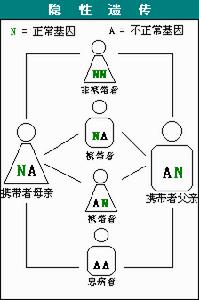 脊髓性肌肉萎縮
脊髓性肌肉萎縮病理變化主要位於脊髓前角,其運動細胞明顯減少,呈退行性變,殘留的神經細胞呈固縮、核溶解,脊髓前根軸突變細,軸突外周細胞腫脹。腦幹運動神經核變性,以面神經、迷走神經、舌下神經多見。肌肉病理檢查見下述輔助檢查部分。
症狀體徵
 脊髓性肌肉萎縮
脊髓性肌肉萎縮1.SMA-Ⅰ型 亦稱Werdnig-Hoffmann 病。約1/3 病例在宮內發病,其母親可注意到胎動變弱。半數在出生1 個月內起病,幾乎所有病例均在5 個月內發病。發病率約為1/10000 出生嬰兒,男女發病相等。多於出生後不久即表現肌張力低下,肌無力以四肢近端肌群受累為主,軀幹肌亦無力。患兒吸吮及吞咽力弱,哭聲低微,呼吸淺,可出現胸廓反常活動。翻身及抬頭困難。腱反射消失。觸診可發現四肢肌萎縮,但常被皮下脂肪掩蓋。眼球運動正常。括約肌功能正常。可見舌肌萎縮和束顫。10%病例可有關節畸形或攣縮。本型預後差。約95%死於出生後18 個月。
2.SMA-Ⅱ型 發病較SMA-Ⅰ型稍遲,通常於1 歲內起病,極少於1~2 歲起病。發病率與SMA-Ⅰ型相似。嬰兒早期生長正常,但6 個月以後運動發育遲緩,雖然能坐,但獨站及行走均未達到正常水平。1/3 以上患兒不能行走。20%~40%患兒10 歲以前仍具行走能力。多數病例表現嚴重肢體近端肌無力,下肢重於上肢,而呼吸肌、吞咽肌一般不受累。有1/3 病例面肌受累。50%以上病例可見舌肌及其他肌肉纖顫。腱反射減弱或消失。本型具有相對良性的病程,多數可活到兒童期,個別活到成年。
3.SMA-Ⅲ型 又稱Kugelberg-Welander 病。一般於幼兒期至青春期起病,而多數於5 歲前起病。起病隱襲,表現為進行性肢體近端肌無力和萎縮。早期大腿及髖部肌無力較顯著,以致病孩行走呈鴨步,登梯困難,逐漸累及肩胛帶及上肢肌群。腦神經支配的肌群通常未受累及,但可出現面肌、軟齶肌無力。眼外肌正常。約1/4 病例伴發腓腸肌假性肥大,此幾乎均見於男性患者。半數患者早期可見肌束顫動。弓形足亦可見到。腱反射減弱或消失。感覺正常。本型預後良好,尤其是女性患者。生存期通常能達到成年,許多患者能有正常壽命。表現較嚴重病例往往為男性患者。本型血清CPK 可有不同程度增高。EMG 除呈神經源性改變外尚可與肌源性損害混雜存在,因此須注意與肌營養不良症鑑別。
4.SMA-Ⅳ型 統稱成年型SMA。發病年齡為15~60 歲,多見於35 歲左右。起病和進展均較隱襲,但亦有呈進行性加重或相對靜止的病例報導。本型預後相對良好,行走能力常可保持終生。發病率小於0.5/10 萬。本型中約1/3 病例呈常染色體顯性遺傳,表現為近端肌無力,進展速度稍快,約5 年後喪失跑步能力。尚有常染色體隱性遺傳類型,則一般表現更加良性病程。另一種類型為X-連鎖隱性遺傳,又稱脊髓腦幹型SMA(Kennedy 病),其發病年齡不等,但常於40 歲前發病。早期表現痛性肌痙攣,可先於肌無力前數年出現。近端肌無力常從下肢開始,逐漸波及肩胛帶肌、面肌及延髓支配諸肌。下面肌及舌肌可見束顫。數年後可出現吞咽困難及吶吃。約50%病例合併一些內分泌功能障礙,表現男性乳房女性化及原發性睪丸病變。
5.其他類型SMA
(1)遠端型SMA:本型約占SMA 的10%,為常染色體顯性或隱性遺傳形式。前者於20 歲前發病,後者稍遲,且症狀較輕。多數患者表現緩慢進展的下肢遠端肌無力和萎縮,脛骨前肌和腓骨肌群尤易受累。弓形足和脊柱側彎亦較常見。約半數病例上肢遠端遲早也會受累,但程度較輕。無感覺障礙。周圍神經傳導速度正常。
(2)慢性不對稱型SMA:本型於16~45 歲起病,男性患者為女性患者的兩倍。表現一個或多個肢體不對稱性肌萎縮,而無錐體束或延髓受累。肌無力可以近端或遠端為主,起病時相對局限於單個肢體。本型自然病程較長,甚至超過30 年。
(3)肩胛腓型SMA:發病年齡30~40 歲。表現肩胛帶肌及下肢遠端肌(尤其是腓腸肌)明顯無力和萎縮。弓形足也較常見。
(4)單肢型SMA:日本和印度曾報導一些病例,其發病年齡各異,男性多見。起病相對較快,而後進入非進展期。由於局限性前角細胞受損,多表現單臂明顯肌萎縮。EMG 顯示嚴格限制於單個肢體的異常。延髓肌及其他肌肉不受侵犯。日本文獻中稱青年型單肢SMA 為平山(Hirayama)病。
(5)此外尚有延髓SMA 並發耳聾(Viatetto-Vanlaere 綜合徵)、兒童延髓型SMA(Fazio-Londe 綜合徵)、眼咽型SMA、面肩肱型SMA、氨基已糖苷酯酶A 缺陷等少見類型。
