疾病概述
 先天性肛門直腸畸形
先天性肛門直腸畸形早在古代,人們對肛門直腸畸形就有了認識,但直至7世紀才有人用細長小刀切開會陰部及腸腔,並用探條擴張治療該畸形。中國在16世紀,明代孫志宏的著作《簡明醫殼》中對肛門閉鎖的手術治療有詳細記載;“罕有兒初生無谷道大便不能者,旬日後必不救,須用細刀割穿,要對孔親切,開道之後,用絹帛卷如小指,以香油浸透插入,使不再合,傍用生肌散敷之自愈。”
18世紀後半葉有人主張在會陰部手術不成功時行結腸造瘺。1835年Amussat用會陰部切開法,強調充分游離直腸,無張力的將直腸黏膜與皮膚縫合的重要性。以後有人為達到充分顯露高位直腸盲端及尿道瘺,而切除尾骨或部分骶骨。19世紀末(1880)NeilMcleod提出腹會陰聯合手術會陰部不能暴露直腸時可從腹部切口游離直腸。直至1948年RhoadsPiper、Randall成功地實施了一期腹會陰手術
20世紀60年代來隨著人們對維持排便功能的神經肌肉的解剖生理肛門直腸畸形、肛周肌肉病理改變的深入研究,對肛門直腸畸形手術方法的改進日趨完善合理,術後肛門排便功能恢復較好。20世紀60年代Stephens強調恥骨直腸肌在維持肛門直腸畸形術後排便功能上的重要性提出對高位畸形行骶會陰或腹骶會陰肛門成形術,即從骶部切口游離已向前上方移位的恥骨直腸肌使直腸盲端經恥骨直腸肌環拖出,以獲得良好的術後排便控制。基於肛門直腸畸形時,外括約肌發育也不正常,肌纖維走向改變,1980年DeVires和Pena提出由骶尾部正中作後矢狀切口將橫紋肌複合體(包括恥骨直腸肌和肛門外括約肌)肌纖維從正中分開然後將直腸置於橫紋肌複合體之中形成肛門,這樣不但能利用恥骨直腸肌,而且也充分利用了外括約肌。近年來不少學者發現肛門直腸畸形,特別是高、中位畸形時,直腸遠端及瘺管處腸壁環肌局限性增厚,即有內括約肌或已具有內括約肌雛形。因此,強調在行肛門成形術時也應儘量保留和利用肛門內括約肌。
因此,治療肛門直腸畸形特別是高、中位畸形的手術原則應是利用電刺激及顯微外科技術,儘量保護和利用那些位置異常和發育不全的肛周肌肉—恥骨直腸肌、肛門外括約肌及肛門內括約肌,使其儘量恢復與直腸之間的正常解剖關係,即一方面應使直腸通過或位於恥骨直腸肌環及外括約肌中心另一方面也應儘量保存和利用肛門內括約肌及其功能。
肛門直腸畸形的首次手術很重要,如處理不當或出現嚴重併發症,不但給再次手術造成困難,更重要的是將明顯影響治療效果。
對肛門直腸畸形的治療不僅要挽救患兒的生命,提高存活率,而且要提高生活質量即要具有正常的排便功能,又能像正常人一樣的生活、學習工作以及參加社會活動為達到此目的除手術治療外正確的術後處理,堅持擴肛以及採用包括微機圖像生物反饋療法的排便訓練等措施也非常重要。
流行病學
據文獻報導,肛門直腸畸形的發生率為1500~5000名新生兒中有1例。國內有關本病的統計不多,上海某醫院婦產科統計30525名新生兒中發現11例,平均為2800∶1。
病因
 先天性肛門直腸畸形
先天性肛門直腸畸形肛門直腸畸形的發生是胚胎髮育發生障礙的結果。引起肛門直腸發育障礙的原因尚不清楚,近年來許多學者認為與遺傳因素有關。根據文獻報導,肛門直腸畸形有家族發病史者在1%以下。佐伯守洋報導的350例肛門直腸畸形中,有3例家族同胞兄弟患同一疾病。VanGelder報導在一個家族中,兄弟姐妹4人均患此病,第1子為肛門狹窄,第2子為直腸尿道瘺,兩個女兒均為直腸陰道瘺。矢野博道等收集29篇文獻中有34個家族發病,與遺傳有關者19組,16組為常染色體隱性或顯性遺傳,3組為伴性隱性遺傳,其中雙胎和三胎者13組,占1/3。也有人認為肛門直腸畸形病兒的同胞中發生該畸形的可能性為25%。
古川敏記對豬的先天性肛門直腸畸形病因調查結果也證明其與遺傳有關。在122頭豬肛門直腸畸形中,因公豬發病的28頭(23%);因母豬發病的6頭(4.9%);因公豬母豬雙方發病者為28頭(32%);原因不明的為60頭(49.2%)。故因公豬、母豬及雙親而發病的共62頭,占50.8%,該學者認為可能在肛門直腸畸形患豬體內有隱性的遺傳因素。1992年有人對家族發病者的發病基因進行研究,發現肛門直腸畸形與位於第6號染色體短臂的HLA有關,認為該畸形的致病基因位於HLA基因附近。張志波研究發現肛門直腸畸形病兒HoxA-13第二外顯子保守區電泳帶型異常,而HoxD-13第二外顯子PCR-PstⅠ限制性內切酶完全未被酶切或酶切不完全。提示HoxA-13、HoxD-13基因異常可能是肛門直腸畸形的致病基因之一有人發現肛門直腸畸形出現在正常家鼠的SD基因突變型鼠,稱此鼠為SD鼠。SD基因以半顯性方式遺傳,影響直腸、泌尿生殖系統中軸骨骼系統的發育。後來有人用雜合子SD鼠繁殖出肛門直腸畸形鼠仔,說明SD基因與肛門直腸畸形有密切關係。
肛門直腸畸形的發生和其他畸形的發生一樣可能與妊娠期,特別是妊娠早期(4~12周)受病毒感染、化學物質、環境及營養等因素的作用有關。胚胎期發生髮育障礙的時間越早,所致畸形的位置越高,越複雜。
1974年Schwetz等人利用Spraque-Dawleg雌性大白鼠於妊娠期間(孕6~15天)吸入不同劑量的氯仿,觀察鼠仔的致畸情況,結果吸入體積分數為100×10-6的氯仿7h,鼠仔除發生缺尾或短尾、皮下水腫、肋骨缺如及胸骨鈣化延遲外,部分鼠仔產生肛門直腸畸形。
1990年Hirai等用乙烯硫脲使妊娠11天的Wistar大白鼠產生肛門直腸畸形胎仔,且畸形類型與人類極相似;我國馮畹蘭、劉穎等也先後用該方法製成肛門直腸畸形的動物模型說明致畸物質——乙烯硫脲是某些胚胎期動物發生肛門直腸畸形的直接原因。
橋本良造給妊娠雌性鼠腹腔內注射視黃酸(all-transretinoicacid)也使鼠仔產生肛門直腸畸形,以高位畸形最多。
上述實驗結果提示,套用致畸物質不但可產生肛門直腸畸形胚仔畸形發生率高達30%~90%也為製作其他各種畸形動物模型展示了良好前景。
發病機制
 先天性肛門直腸畸形
先天性肛門直腸畸形1.胚胎髮生在胚胎第3周末,後腸末端膨脹與前面的尿囊相交通形成泄殖腔。中腎管(午菲管)—原腎管開口於泄殖腔中。泄殖腔的尾端被外胚層的一層上皮細胞膜所封閉,稱為泄殖腔膜,使與體外相隔。胚胎第4周,位於泄殖腔與後腸間的中胚層皺襞形成並向尾側生長;同時,間充質於泄殖腔兩側壁的內方增生形成皺襞,向腔內生。它們構成尿直腸隔將泄殖腔分為前後兩部分,前者為尿生殖竇,後者為直腸,使兩個系統的交通越來越小,逐漸形成一個小管道,稱為泄殖腔管,於胚胎第7周時完全封閉。尿直腸隔由兩個內胚層板構成(尿生殖層和直腸層)在兩層之間充滿中胚層組織和生殖胚芽。
尿直腸隔與泄殖腔膜的中央處融合,並向外突出成為會陰矩狀突—未來會陰的胚芽。同時泄殖腔膜也被分為前後兩部分,前者為尿生殖竇膜後者為肛膜胚胎第7~8周時,兩個膜先後破裂。肛門的出現不僅由於肛膜破裂,在此以前從胚胎第5周開始,外胚層向肛膜的外表面發展,形成肛凹,肛凹逐漸加深接近腸管,肛膜破裂使起源於外胚層的肛凹與內胚層發生的直腸相通。
胚胎第4個月會陰向前後方向迅速增長,因此使肛門後移至通常位置。生殖器官和會陰的形成與上述過程同時進行。在女胎內生殖器官由米勒管形成,該管開始與中腎管一起發展,向下延伸至中胚層的尿直腸隔的深部,米勒管的中段和下段靠近並融合在一起形成子宮和陰道其上部沒有融合則形成輸卵管,午菲管退化。
在女胎泄殖腔分隔以後,生殖皺襞的後半部與尿直腸隔的會陰矩狀突癒合在一起形成會陰和叉狀的陰道前庭原基,生殖隆突沒有癒合,變成大陰唇;生殖皺襞的前半部也沒有癒合形成小陰唇。
在女胎泄殖腔形成和分隔期受某種因素或致畸物質的影響出現發育障礙,可構成下列畸形:O-直腸泄殖腔畸形;p-直腸膀胱瘺(米勒管中部未癒合時這種畸形伴有雙角子宮;下部未癒合時伴有雙陰道);q-直腸陰道瘺;r-直腸前庭瘺;s-肛門正常,直腸前庭瘺;t-肛門直腸發育不全,無瘺;u-肛門發育不全,無瘺。
後期發育停止導致出生後病兒肛膜未破(v)。
會陰矩狀突發育不全時,生殖皺襞是形成會陰的基本來源,生殖皺襞肥大,在通過肛管的正常肛穴部位癒合所致的畸形,稱為隱蔽肛門(w)。
前會陰肛門(x)是會陰發育不良肛門沒有後移至正常位置的結果。
在沒有分化性別期泄殖腔的分隔過程在男胎和女胎都一樣其基本差別是在內、外生殖器官和會陰形成時期出現的。午菲管發育成睪丸和中腎管變為輸精管的同時米勒管退化。
在男胎形成會陰時,生殖結節增長形成陰莖。生殖皺襞左右癒合覆蓋於尿生殖竇的表面(圖2fi)形成前部尿道和尿道球部。在生殖皺襞外側的生殖隆突則形成陰囊,沿矢狀線癒合處為陰囊正中縫(圖2k)和女胎一樣,男胎在第4個月以後的發育中會陰迅速向前後方向發展將肛門推移至正常位置(圖2m,n)。
肛門直腸畸形的發生,男胎和女胎在原則上是相同的,只有解剖特點的區別
泄殖腔分隔障礙的結果,是使尿生殖竇和直腸之間相通,在男孩可出現泄殖腔畸形,而較多見的是直腸泌尿系瘺,瘺管可位於膀胱三角部(直腸膀胱瘺)或尿道前列腺部(直腸尿道瘺)。當瘺管閉塞時出現肛門直腸發育不全無瘺)。
胚胎髮育後期出現發育障礙,結果可形成肛門發育不全,無瘺,肛膜未破,肛膜狹窄。會陰發育不全,可構成前會陰肛門和肛門皮膚瘺、不完全性隱蔽肛門。
胎兒直至出生時直腸仍呈紡錘狀,上端球狀膨脹部稱肛球,相當於成人的直腸壺腹部,紡錘狀管以下另有一短而不明顯的膨大部,稱尾球,相當成人的直腸肛門部的下部。尾球存在的時間較短,第8周時大部已基本消失。肛門直腸正常的直腸閉鎖,往往發生在肛球上端,相當於肛門上3~4cm處,可能與胚胎性狹窄有關。
會陰部肌肉是就地發育的,它起源於會陰部間質,在胚胎第2個月時已存在皮肌的形態,稱泄殖腔括約肌。第3個月時皮肌分化為肛門外括約肌、肛提肌和尿生殖竇括約肌,當生殖器官形成後(第4、5月),尿生殖竇括約肌又分出膜部尿道括約肌、坐骨海綿體肌、會陰淺橫肌等,以後再分出會陰深橫肌。肛門直腸畸形病兒上述各肌雖然存在,但在高中位畸形時,外括約肌和肛提肌有不同程度的改變。
2.病理類型先天性肛門直腸畸形的分類方法很多,名詞術語也不統一文獻中對這些畸形的記載混亂,很難對比不同分類的治療效果。
(1)Ladd-Gross分類法:過去在我國多採用Ladd和Gross於1934年提出的4型分類法,即第1型肛門或直腸下端狹窄;第2型肛門膜狀閉鎖;第3型肛門閉鎖,直腸盲端距皮膚有相當距離;第4型直腸閉鎖。以後又將第3型分為高位和低位2型這種分類方法是單純從解剖形態上制定的,對手術方法和途徑的選擇以及預後的估計均無重要意義。
(2)國際分類法:1970年在澳大利亞召開的國際小兒外科醫生會議上,制定了高位、中間位和低位的分類方法,它以該畸形的胚胎髮生和病理改變為基礎對指導臨床實踐和估計預後均有幫助,是目前較合理的分類方法該分類法是對許多分類方法的折中和修訂已被各國小兒外科醫生廣泛套用。
國際分類法的主要特點是以直腸盲端與肛提肌特別是恥骨直腸肌的關係做為區分高、中、低位的標準,即直腸盲端終止於肛提肌之上者為高位畸形;直腸盲端位於恥骨直腸肌之中被該肌所包繞為中間位畸形;穿過該肌者為低位畸形。Stephens發現在肛門直腸畸形病兒的恥骨直腸肌位置有改變,強調在做肛門成形術時注意保護該肌並使直腸通過該肌環,對決定術後肛門排便功能有重要性。其次,國際分類提出了介於高低位之間的移行型,即中間位畸形,而這種畸形大部分應行骶-會陰肛門成形術,對合理選擇術式有指導作用
(3)wingspread分類法:為便於套用1984年將國際分類法種加以簡化,修改後的分類法稱為Wingspread分類法,具體分類如下:
 先天性肛門直腸畸形
先天性肛門直腸畸形男性:
①高位:A.肛門直腸發育不全:a.直腸前列腺尿道瘺:瘺管開口於後尿道,無肛門內括約肌,外括約肌不明顯,盲端位於PC線上。b.無瘺:盲端與尿道間可有纖維索帶連線,無肛門內括約肌,僅有外括約肌痕跡,盲端平或高於PC線。B.直腸閉鎖:直腸盲端止於不同高度肛門及肛管正常有肛門內、外括約肌及肛提肌,且與肛管保持正常關係。
②中間位:A.直腸尿道球部瘺:直腸盲端位於尿道球部海綿體肌之上,恥骨直腸肌包繞直腸盲端瘺口,肛門內括約肌缺如,直腸盲端位於PC線與Ⅰ線之間。B.肛門發育不全,無瘺:直腸盲端終於尿道球部海綿體肌之上,恥骨直腸肌環繞直腸盲端。肛門內括約肌缺如,外括約肌僅見痕跡,直腸盲端位於PC線與Ⅰ線之間。
③低位:A.肛門皮膚瘺:瘺管開口於會陰部至陰囊縫線或陰莖腹側的任何部位以陰囊部居多。肛管呈瓣狀瘺管被菲薄的皮膚掩蓋。恥骨直腸肌正常。B.肛門狹窄:肛門及內、外括約肌正常。
除上述3類畸形外,還有一些罕見畸形
女性:
①高位:A.肛門直腸發育不全:a.直腸陰道瘺:直腸盲端開口於陰道後壁中部b.無瘺。B.直腸閉鎖
②中間位:A.直腸前庭瘺:直腸盲端位於PC線上或稍下,瘺管長1~2cm,通過恥骨直腸肌沿陰道後壁開口於陰道前庭部。B.直腸陰道瘺:瘺管開口於處女膜上方,恥骨直腸肌環繞直腸盲端與瘺管。C.肛門發育不全,無瘺:直腸盲端終於陰道下端平面,尿道及陰道正常直腸盲端位於Ⅰ線或其下。
③低位:A.肛門前庭瘺:瘺管甚短直腸與陰道緊密相鄰恥骨直腸肌正常有肛門內括約肌痕跡肛門外括約肌有時存在,瘺口位於陰道前庭部瘺口周圍為黏膜。B.肛門皮膚瘺。C.肛門狹窄。
④泄殖腔畸形這是一種較少見的肛門直腸畸形,即直腸、陰道、尿道共同開口在一個腔,一般按Raffenspersgers分型法分型由於該分型法過於複雜為便於套用,有人按病理解剖特點將其分為3種類型
A.常見型:共同管長2~3cm,陰道大小正常,肌肉複合體及肛門外括約肌位置正常。
B.高位型:共同管長3~7cm,骶骨發育短小,肌肉發育薄弱,陰道狹小,骨盆前後徑小,一般術後效果不理想。
C.低位型:共同管長0.5~1.5cm,又稱低位直腸陰道瘺合併女性尿道下裂,盆部發育正常,預後較好。本病常合併雙陰道、雙子宮,約占60%巨大陰道積液約占40%。
⑤罕見畸形
3.病理改變20世紀70年代,不少學者對肛門直腸畸形病兒的盆腔結構進行解剖組織學研究,證明該畸形不僅肛門直腸本身有閉鎖和發育不全,同時盆底肌內、骶骨、神經及肛周皮膚等均有不同程度的病理改變,肛門直腸畸形的位置越高,這種改變越明顯,越嚴重。
(1)肌肉改變:
①恥骨直腸肌:Stephens對29例肛門直腸畸形病兒屍體進行解剖,發現2例高位肛門直腸畸形的男嬰,恥骨直腸肌依附於尿道後壁;1例直腸陰道瘺者,該肌附著於陰道後壁並向前移位;而在患前庭瘺和肛門閉鎖的病例中,該肌處於正常位置。Stephens指出恥骨直腸肌的發育與骶椎缺如有關,如骶2以下缺如,該肌不發育;骶3以下缺如,該肌發育薄弱;骶4以下缺如,該肌可正常發育。Kiesewetter曾做9例解剖,強調該肌有上移即高位畸形時,恥骨直腸肌處於恥尾線(PC線)水平;而低位畸形該肌遠離PC線。王常林觀察和測量該類畸形病兒恥骨直腸肌的位置和長度(表2),發現高位畸形時恥骨直腸肌上、下緣延長線與恥尾線的交角明顯小於正常兒。其後上、下緣與肛穴的距離明顯大於正常兒,說明該肌上移,另外恥骨直腸肌的長度較正常兒短。恥骨直腸肌與外括約肌分離與骶椎間隙增大,由脂肪占據。中位畸形時恥骨直腸肌雖有上移和縮短但不如高位者明顯,與正常兒比較無顯著差異該肌纖維包繞直腸盲端,且直腸盲端位置越低,被肌纖維包繞的越多。該肌在直腸盲端的後外方與外括約肌深淺部肌纖維相接。直腸前庭瘺者和低位畸形一樣,恥骨直腸肌環繞於直腸或瘺道的後方,處於正常解剖位置。
總之,肛門直腸畸形病兒的肛提肌包括恥骨直腸肌的發育良好,僅個別病例該肌缺如或發育不良。由於畸形類型不同,恥骨直腸肌的位置可發生改變,即高位畸形該肌明顯向上向前移位並短縮呈閉鎖狀,依附於前列腺尿道或陰道後方,並與直腸盲端和外括約肌有一定距離。因此高位畸形行肛門成形術時,應設法使直腸準確地通過恥骨直腸肌環。中位畸形時直腸盲端位於恥骨直腸肌之中,被該肌所包繞,其肌纖維與外括約肌纖維相連。直腸前庭瘺和低位畸形恥骨直腸肌環繞於直腸後壁基本處於正常位置。
②外括約肌:胚胎研究證明,外括約肌是單獨發育的,與肛門直腸畸形的發生無關Kiesewetter報導,肛門直腸畸形病兒存在外括約肌,在臨床上有人用電刺激或肌電圖研究觀察,也證明了這一點。Smith在16例病兒中,經組織切片觀察發現1例外括約肌缺如,另1例外括約肌前部缺如。Stephens也看到2例直腸尿道瘺的病兒無外括約肌也有人認為高位畸形時外括約肌發育不良或僅為痕跡器官。
王常林對16例肛門直腸畸形患兒的盆腔正中矢狀斷面標本進行解剖和組織學研究,證明外括約肌均存在,由於畸形類型不同,該肌的分布、形態大小和肌纖維走行方向變化較大。用方格圖表法對13例盆腔完整標本的外括約肌分布面積進行對比觀察,並與6例正常新生兒標本對照,發現肛門直腸畸形病兒外括約肌的面積較正常兒增大,即低位畸形時外括約肌面積與正常兒基本一致;中位畸形時為正常兒的1.4倍;高位畸形時,僅1例外括約肌明顯縮小,約為正常的1/2,並移位至尾骨尖部,其餘均較正常兒的面積大,平均為正常兒的2.5倍,在外括約肌內部有不同程度的脂肪充填。
在正常兒盆腔正中矢狀斷面上,肉眼觀察外括約肌呈前、後兩團,位於肛管的前後方。肛門直腸畸形病例直腸盲端的位置越高,兩團結構越不明顯,不易分開甚至失去正常形態。在鏡下觀察外括約肌纖維走行方向正常兒外括約肌深淺部肌纖維呈橫斷面,低位畸形其肌纖維也為橫斷面;而中位畸形肌纖維多為斜行,僅少部分為橫斷面;高位畸形時深淺部肌纖維多為斜行及縱行呈橫斷面者甚少有的病例幾乎以縱行肌纖維為主,呈高柱狀,總之外括約肌肌纖維走行方向異常紊亂。
張志波等(1997)對肛門直腸畸形動物模型的肛門外括約肌進行組織化學觀察發現,在高、中位畸形的單位面積中無論是肌纖維數,還是收縮,慢耐疲勞的Ⅰ型肌纖維所占比例均明顯減少,而低位畸形則基本正常我們對肛門直腸畸形肛門外括約肌的超微結構進行觀察發現部分肌原纖維排列紊亂,結構不清,有的呈溶解狀態;Z帶有不規則改變,扭曲、斷裂;線粒體大小不等,嵴有缺失、斷裂空泡變,有早期髓鞘樣變,這些改變可能與該畸形有骶髓和肛周組織中神經發育不良有關。
於明等採用體表電極肌電圖技術,對32例肛門直腸畸形(高位7例中位12例,低位13例)病兒肛門外括約肌肌電活動進行檢測觀察肛穴處靜止時波幅與頻率及刺激時波幅與頻率4項肌電活動指標結果高位畸形有2項明顯低於正常,中位有1項,而低位畸形肌電活動基本正常。從功能上證實肛門直腸畸形病兒無論高中、低位畸形均存在肛門外括約肌,但直腸盲端位置越高,外括約肌發育越差。在肛穴的不同部位檢測肌電活動結果表明肛門直腸畸形兒肛門外括約肌肌電活動最強處不一定在正常肛穴位置有11例(31%)位置偏前偏後或偏側,以高位畸形偏位者最多中間位其次,低位又次之,各型又以向前偏位者多見。因此,對肛門直腸畸形病兒術前利用肌電圖檢查,確定肛門外括約肌的發育程度位置及範圍術中儘量辨認和保存括約肌,不僅使直腸盲端通過恥骨直腸肌環,而且要穿過外括約肌中心是十分重要的。
③內括約肌:關於肛門直腸畸形病兒有無內括約肌文獻中說法不一Stephens和Kiesewetter等認為肛門直腸畸形病兒無肛管,也無內括約肌。Scott(1959)在直腸前庭瘺病兒發現內括約肌存在。秋山洋(1973)和Nixon(1976)等證明,在低位肛門畸形時內括約肌存在。就是在1984年修定的肛門直腸畸形國際分類也標明:高位和中位畸形內括約肌缺如,低位畸形內括約肌存在但早在1958年Bill就發現伴泌尿生殖系瘺的高、中位肛門直腸畸形有內括約肌,並認為畸形的發生是直腸移行過程中受抑制而停滯於膀胱尿道或陰道,未達到正常位置的結果。Gans(1961)對肛門直腸畸形進行病理研究,其結果與Bill的觀察一致。王常林等(1983)報導了對10例肛門直腸畸形完整病理標本的組織學研究,在5例高位畸形標本中,3例於直腸遠端腸壁環肌層有局限性增厚,範圍較小;4例中位畸形中3例也有局限性環肌增厚,範圍稍大,多為前後兩處,另1例前庭瘺其內括約肌發育良好;1例低位畸形內括約肌基本正常。
故此他們認為肛門直腸畸形病兒內括約肌的發育程度與畸形類型有關,即位置越高,發育越差,甚至完全缺如。
1987年Lambrcht報導對33隻新生仔豬肛門直腸畸形的形態學觀察結果:公豬24隻,母豬9隻,高位畸形18隻,低位畸形15隻。所有動物均見有內括約肌環繞於瘺的近端,局限於瘺進入直腸盲端的開口部位。內括約肌的形態差異很大,有的像正常的內括約肌一樣呈漏斗狀;但大部分內括約肌分布較寬,呈圓盤狀或平碟狀。在肛門畸形瘺的近端有肛管的很多特徵:A.被內括約肌環繞;B.在內括約肌部腸壁內神經節細胞減少或缺如;C.瘺的近端被有移行上皮;D.內有肛門腺1990年Rintala對10例高、中位肛門直腸畸形病兒的直腸盲端、尿道瘺、會陰瘺進行組織學檢查,發現9例有正常肛管移行上皮,在該區域內為低神經節細胞區,且膽鹼酯酶呈強陽性反應。
劉穎等(1996)對28隻肛門直腸畸形鼠肛門內括約肌觀察發現有瘺型肛門直腸畸形其瘺管內覆有未角化的復層上皮,在該處直腸末端環肌層明顯增厚,肌細胞發育正常。即有瘺型肛門直腸畸形具有明顯的內括約肌,無瘺型肛門直腸畸形直腸盲端內無復層上皮,環肌也未增厚即無內括約肌;而無瘺型低位畸形直腸盲端覆有角化鱗狀上皮,範圍較廣,在有鱗狀上皮的範圍內環肌層局限性增厚明顯,肌細胞發育正常即存在內括約肌。
另外,也有人研究證明,在肛門直腸畸形病兒的瘺管處具有內括約肌功能。Ohama(1990)在術前由結腸造瘺口插入導管測量高中位畸形瘺管處的壓力,當擴張直腸時,該處壓力下降,即有正常肛管直腸反射。該反射反映肛門排便控制系統的完整性和神經肌肉的協調性,內括約肌的完整性是該反射存在的關鍵條件。
肛門內括約肌通常處於持續收縮狀態,維持肛管高壓,並構成肛管與直腸間的壓力屏障,為控制排便的重要因素之一有人認為內括約肌全部切除,肛管靜止壓力約下降50%。
上述資料說明多數肛門直腸畸形(包括高中位畸形在內)病兒都有內括約肌。因此,應行保留內括約肌的肛門成形術,即手術時保留直腸盲端及瘺管,這樣可以最大限度地保存儘管是發育不全的內括約肌,以便獲得較好的排便功能。自1982年以來李正等採用保留內括約肌的肛門成形術對19例高位畸形病人術後隨訪6.5年肛門功能臨床和綜合評定為優者分別由1982年前的23%、26.9%提高到63.2%和57.9%。Husberg1992年報導對48例高中位肛門直腸畸形採用保留內括約肌的肛門成形術的隨訪結果,對4歲以上的22例進行直腸肛管測壓15例直腸肛管反射正常;其中11例排便功能正常,而6例該反射消失者均有污糞。
④腸壁縱肌:在部分高、中位畸形病例中,可見直腸盲端腸壁縱肌向下延伸,可延伸至外括約肌的肌纖維內,其長短不同。
(2)神經改變:
①骶髓改變:李龍等(1993)報導對10例肛門直腸畸形兒的骶髓標本進行觀察,其中高位畸形4例,中位1例,低位5例。病兒末段骶髓均存在異常改變其中6例標本的中央管呈菱形擴大,實質變薄;1例從第4骶髓節段以遠,中央管和前正中裂未發育,左右前角內側群的運動神經元在中線處融合;1例低位畸形末段中央管內有一矢狀走行隔膜;另外2例,末段骶髓的中央管橫向擴大,似脊髓裂樣改變。
畸形標本中,有2例第4和第5及其以遠的骶骨缺如,但是其相應的神經根卻存在,並分別穿出硬脊膜
除上述形態改變外先天性肛門直腸畸形兒骶髓前角內側群的運動神經元的數目較正常兒減少,高、中位畸形和低位畸形分別為正常的34.4%和70.5%。
目前已明確,神經管為胚胎早期發育的中軸器官,它誘導附近各胚層結構的分化和發育。肛門直腸畸形兒末段骶髓的異常改變,可能意味著此畸形在胚胎早期,因尾端神經管發育異常,致盆底和會陰部組織發育畸形。
骶髓前角內側群的運動神經元是盆底肌肉和肛門外括約肌的運動神經中樞,肛門直腸畸形兒此群運動神經元數目減少,與其周圍神經的改變一致。
②骶神經改變:肛門直腸畸形病兒常伴有骶椎畸形。當骶椎椎體缺如時,可伴有骶神經的改變,缺如的節段越多,骶神經改變越明顯。Smith解剖6例肛門直腸畸形兒屍體,2例第2骶椎以下缺如,未見骶神經及會陰神經;2例第3骶椎以下缺如,骶神經僅有3對,這4例骶神經缺如數目與骶骨缺如節段相一致,稱為預料型。另2例第2骶椎以下缺如者1例左側沒有骶神經,而右側有7支骶神經發出,兩側會陰神經都來自於右側骶神經;1例骶神經分別從第4、5腰椎之椎間孔、第5腰椎和第1骶椎之椎間孔發出,兩側會陰神經都存在。這種骶神經改變與骶骨缺如節段不一致,稱為非預料型。
王常林等(1992)報導16具肛門直腸畸形兒屍體(高位畸形10例,中間位畸形6例)解剖發現:其中14例患兒解剖顯示第2、3、4骶神經存在該神經從相應椎孔發出後,沿椎體兩側下行有分支到達盆底肌肉;在僅有3個骶椎的病例,硬脊膜終止於第3骶椎下方1.5cm處在其最低處有一束馬尾神經穿出,形成第4對骶神經;在第23、4椎體融合的病例,相當於第3椎間孔處發出一對骶神經。
骶神經與直腸盲端的關係:在10例高位畸形中,有5例直腸盲端位於第2骶椎水平以上,第2、34骶神經與直腸盲端無聯繫;其餘5例,直腸盲端在第2骶椎水平以下者,1例第2骶神經進入直腸壁;3例第3、4骶神經,1例第2、34骶神經與直腸壁相連。中間位畸形6例,直腸盲端均與骶神經有聯繫其中4例與第3、4骶神經相連,2例與第23、4骶神經相連。
肛門直腸畸形兒骶椎有明顯改變者,可伴有骶神經的發育異常直接影響本病的治療和預後。
據Ленюшкин報導在肛門直腸畸形術後排便功能障礙的病例中約有10%的病例是骶椎畸形,神經發育障礙所致因而臨床上觀察畸形兒骶椎改變具有重要意義。
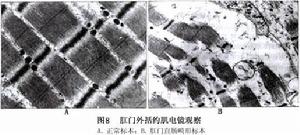
③肛周組織中神經末梢改變:在正常兒盆底及肛周組織中共有4種感覺神經末梢存在:肌梭,位於恥骨直腸肌的前2/3段內和肛門外括約肌的中段內;環層小體位於內括約肌與外括約肌之間的組織中和骶前間隙內;球樣末梢,位於骶前間隙內;游離神經末梢,分布於肛管的黏膜上皮和肛周皮膚中。
李龍等報導11例死於新生兒期的先天性肛門畸形兒(高位5例,中位5例,低位1例)盆底及肛周組織中的感覺神經末梢形態學觀察結果發現肌梭僅見於恥骨直腸肌中,分布在中1/3段內;在肛門外括約肌中未見肌梭,且發育不良。
李正等發現,高位和中位肛門畸形兒恥骨直腸肌、肛門外括約肌和骶前間隙內的感覺神經末梢呈發育不良改變一方面,感覺神經末梢的密度降低以肛門外括約肌和骶前間隙內的感覺神經末梢降低為甚,同時儘管恥骨直腸肌中的肌梭數較正常兒少,但是確實存在有一定數量的肌梭;另一方面,骶前間隙內感覺神經末梢發育不良。
目前已經明確,肌梭、環層小體和球樣末梢分別為牽張反射、壓力感覺和溫熱感覺的感受器。許多學者認為正常人恥骨直腸肌和肛門外括約肌中的肌梭是構成該肌肉在一般狀態下持續收縮反射和擴張直腸時肛門外括約肌收縮反射的感受器,同時它與肛周組織中的環層小體、球樣末梢觸覺小體等共同參與便意產生過程。
Kiesewetter在對高、中位肛門畸形兒術後複查時發現,刺激排便控制功能較好患兒的恥骨直腸肌區,可產生排便感。這可能是刺激興奮了此肌肉中肌梭的結果。恥骨直腸肌中的肌梭是高、中位畸形兒重要的排便感受器。
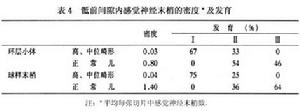
正常新生兒恥骨直腸肌和肛門外括約肌中運動神經末梢的分布:運動終板分布於恥骨直腸肌的中段內和肛門外括約肌的兩側段內。兩肌肉中,平均每張切片運動終板的數目分別為133.58±76.84和37.74±13.53。恥骨直腸肌和肛門外括約肌中平均每張切片神經束的數目分別為94.06±45.43和66.16±32.82。肛門直腸畸形兒恥骨直腸肌和肛門外括約肌中運動神經末梢的分布:運動終板的分布與正常兒相似,但是其面積較正常兒小,且著色淡,高位和中位肛門直腸畸形兒恥骨直腸肌中,平均每張切片運動終板的數目分別為14.00±11.30和18.31±8.38。肛門外括約肌中,運動終板數目分別為9.57±4.92和10.70±4.57。高中位畸形兒兩肌肉中運動終板的密度較正常兒明顯降低。
高位和中位肛門畸形兒的恥骨直腸肌中平均每張切片中神經束的數目分別為14.29±12.90和21.92±11.06;肛門外括約肌中神經束的數目分別為9.29±7.13和14.67±7.93高、中位肛門直腸畸形兒兩肌肉中神經束的密度較正常兒明顯降低。
運動神經末梢是控制肌肉活動的重要環節高、中位肛門直腸畸形兒恥骨直腸肌和肛門外括約肌中的運動神經末梢呈發育不良改變,其程度與兩肌肉中感覺神經末梢的改變一致。
④直腸遠端腸壁內神經改變:肛門直腸畸形兒直腸遠端腸壁內膽鹼能、肽能和腎上腺能神經也有不同程度的改變。王偉等(1993)對8例無肛畸形兒(高位3例,中位3例,低位2例)直腸遠端腸壁內膽鹼能、肽能和腎上腺能神經分布進行觀察發現:在黏膜下層AchE陽性神經叢及神經節細胞數及肌間的神經叢數較正常兒略有減少,酶活性減弱,而肌間的AchE陽性神經節細胞數則明顯減少並且以不成熟型為多,每個視野面積內僅為1.3個而在正常兒則為2.7個;在黏膜下層和肌間,SP能陽性神經叢和神經節細胞數量也明顯減少,新生兒位於黏膜下層及肌間的神經節細胞數為12.42個和13.28個而肛門直腸畸形兒則僅為2.50個和7.83個。其免疫反應新生兒呈弱陽性者分別為15.9%和7.5%,而肛門直腸畸形兒則高達53.3%和100%。另外在肌間腎上腺能陽性神經纖維較正常兒減少,螢光強度減弱
⑤肛門部皮膚神經改變:正常兒肛門部皮膚有豐富的感覺神經末梢能辨別直腸內容物的性質是固體、液體和氣體因此,許多學者強調行肛門成形術時應充分利用肛穴部的皮膚形成肛管,以保留感覺功能。
Kiesewetter研究肛門成形術後肛門直腸的感覺功能發現,低位畸形行會陰肛門成形術的病例肛門直腸感覺功能良好;而高位畸形行拖出型腹會陰肛門成形術的病例,僅在齒狀線上1~2cm的直腸黏膜有感覺其他部位無感覺認為這種感覺的產生是皮膚感覺神經末梢進入直腸遠端黏膜的結果
Ленюшкин對肛門直腸畸形病例肛穴部皮膚進行組織學研究,發現該處皮膚菲薄,乳頭變平,全部表皮被2~3層細胞和角化層覆蓋,特別是沒有神經纖維和神經末梢像神經切除術後的皮膚組織學改變一樣。肛穴部皮膚發育不良的面積為(0.3~0.5)cm×(1.0~1.5)cm其大小與肛門直腸畸形的位置高低無關。
李正等對11例肛門直腸畸形兒肛門部皮膚進行組織學檢查,結果與Ленюшкин的觀察不一樣,在該處皮膚與皮下組織中均有神經纖維存在,但是高位和中位畸形兒神經纖維的密度明顯低於正常兒,且高位低於中位。
總之,從骶髓到盆腔和肛周組織中各種神經末梢的改變,即神經病理改變也是肛門直腸畸形的重要病理改變,其病理改變程度與畸形類型有關畸形位置越高,其神經病理改變越明顯。
各類肛門直腸畸形病兒盆腔結構改變舉例如下。
例1:男,高位肛門直腸發育不全、無瘺。在盆腔正中矢狀斷面上肉眼可見直腸盲端位於第1骶椎水平,距肛穴皮膚5.0cm恥骨直腸肌明顯向前上方移位、短縮,呈閉鎖狀態,依附於前列腺後方該肌距直腸盲端2.0cm,並與外括約肌分離。外括約肌處於恥骨直腸肌和肛穴之間,其前後方有脂肪充填。
鏡下所見:恥骨直腸肌未顯示。外括約肌深淺部位於尾骨尖前下方的脂肪團塊與前列腺之間,肌纖維基本為縱行走向呈柱狀直達肛穴,將外括約肌皮下部分為前後兩部,該肌靠上方肌纖維排列緻密,靠下方肌纖維排列較疏鬆,肌纖維間有脂肪和結締組織充填。外括約肌皮下部面積較廣,其肌纖維平行皮膚走向。直腸盲端鏡檢腸壁部分環肌有增厚。
另1例高位肛門直腸發育不全,無瘺。在盆腔正中矢狀斷面上肛穴上方被一巨大的梨形脂肪組織所占據,外括約肌明顯向後上方移位,位於尾骨尖的前下方,面積僅為正常外括約肌的1/2。在該肌中部有一條纖維脂肪隔將其分為前下和後上2部。前部與恥骨直腸肌相連肌纖維呈斜斷面及橫斷面;後部與尾骨相連,肌纖維多呈斜斷面。
例2:男,中位肛門發育不全,無瘺在盆腔正中矢狀斷面上可見直腸盲端擴張,位於尾骨尖水平,並受2、3、4骶神經支配。恥骨直腸肌包繞著直腸盲端較菲薄,外括約肌在恥骨直腸肌與肛穴之間。
鏡下所見:恥骨直腸肌包繞於直腸盲端外括約肌深淺部位於恥骨直腸肌下方,中間有脂肪及結締組織將其分為前後2部。前部較小,位於海綿體肌後方肌纖維多呈橫斷面和斜斷面;後部較大,與恥骨直腸肌相連,多呈橫斷面、斜斷面。
直腸盲端腸壁較薄,前壁有一處可見環肌增厚。
例3:女,直腸前庭瘺在盆腔正中矢狀斷面上,直腸不擴張,受2、3、4骶神經支配。恥骨直腸肌繞過直腸及瘺道後方。外括約肌分前後兩團,較易辨認,位於瘺道後方。骶骨有反曲,第2、3、4骶椎融合只顯示一個骨化核。
鏡下所見:恥骨直腸肌環繞於直腸及瘺道後方,外括約肌呈前後兩團,均位於瘺道後方。前團面積小,與瘺道後壁相鄰,肌纖維呈橫斷面;後團面積較大其上方與恥骨直腸肌相連,肌纖維多呈橫斷面部分為斜斷面,皮下部從尾骨尖向下,與皮膚平行走行到肛穴處中止。外括約肌的位置結構基本正常瘺道自直腸遠端向下伸延,該處腸壁環肌已開始逐漸增厚,直達瘺道的遠端,已形成較完善的內括約肌瘺口周圍結締組織緻密。
例4:女,肛門皮膚瘺。在盆腔正中矢狀斷面上,瘺開口於陰唇後聯合稍後方,直腸被一巨大多囊性腫物擠壓向前移位,僅有2個骶椎,第2骶椎椎體明顯變小。
鏡下所見:外括約肌深淺部呈一團與恥骨直腸肌相連,肌纖維多呈橫斷面,皮下部與皮膚平行走向,分布於瘺口後方腫瘤病理診斷:骶前畸胎瘤。
4.伴發崎形先天性肛門直腸畸形經常伴發其他畸形一般報導其發生率為28%~72%。Stephens和Smith在246例肛門直腸畸形中發現149例(60.6%)伴發1種或多種畸形。有人收集3223例肛門直腸畸形,伴發畸形的發生率為43.4%但實際上比上述的數目要多,因為有一些內臟畸形尚未被發現。有人報導屍檢發現伴發畸形為92%。有些病例為多發性畸形約1/5病例伴發嚴重的危及生命的畸形。多數學者一致認為,高位肛門直腸畸形伴發畸形的發生率多於低位畸形而且更嚴重。Cook報導在利物浦醫院219例高中位畸形中,159例(72.6%)伴發其他畸形而在165例低位畸形中,35.2%(58例)伴發其他畸形。在高位畸形中伴發畸形的發生率男女比例基本一樣,而在低位畸形中伴發畸形的發生率女多於男,分別為50%和25%高位畸形中76例(34.7%)死亡,而低位畸形僅20例死亡(12.1%)。二組中死亡病例多數死於其他系統畸形。伴發畸形最多的為泌尿生殖系畸形,其次為脊柱,特別是骶椎消化道、心臟以及其他各種畸形。有人將肛門直腸畸形及其伴發畸形歸納為VATER綜合徵:(V:脊柱、心血管;A:肛門;T:氣管;E:食管;R:腎臟及四肢),並指出某些畸形合併發生的非隨機傾向用圖表示彼此間相對發生的幾率。
 先天性肛門直腸畸形
先天性肛門直腸畸形肛門直腸畸形多伴發泌尿生殖畸形,且多為上尿路複合性嚴重畸形,近年來文獻報導較多。目前不少醫生在發現肛門直腸畸形之後,沒有檢查有無伴發泌尿系畸形以致使大部分並發畸形被漏診,或只到晚期已發生嚴重結構和功能障礙,發展到慢性腎功能不全的程度才確診。
自1952年曾有學者報導120例肛門直腸畸形中有41例(34%)合併泌尿系異常後,文獻中各學者報導的發生率差別較大從19%~77%。1974年宮野等在100例肛門直腸畸形兒中60例進行了IVP檢查58例進行了排尿時膀胱尿道造影檢查,發現上尿路異常32例(53%),下尿路異常16例(26%)在平本等報導的273例肛門直腸畸形中,上尿路異常53例(19.4%),下尿路異常23例(8.8%)。一般上尿路畸形包括單側腎缺如、腎發育不良、孤立遊走腎、融合異位腎、馬蹄腎單或雙側腎積水、巨輸尿管、膀胱輸尿管反流等,以單側腎缺如、腎發育不良和膀胱輸尿管反流較常見下尿路畸形包括神經膀胱、膀胱外翻、尿道狹窄、尿道下裂等。
Атакулов(1983)在186例肛門直腸畸形中發現有81例(44%)伴發泌尿生殖畸形其中上尿路畸形48例,包括腎發育不全、腎盂輸尿管積水、雙腎雙輸尿管畸形輸尿管開口異位、輸尿管囊腫等;下尿路畸形53例,包括神經膀胱、直腸尿道瘺、尿道狹窄、憩室和重複畸形等;生殖系統畸形16例,如尿道下裂、泄殖腔畸形、隱睪等Атакулов最初只對有泌尿系症狀的肛門直腸畸形病例進行泌尿系檢查,結果在124例中發現34例(27%)泌尿系畸形,以後對肛門直腸畸形病例常規做泌尿系檢查,在61例中發現47例(占77%)泌尿系畸形,較前期增加2倍。因此,他強調對每個病人都應常規進行泌尿系檢查同時還發現男孩伴泌尿系畸形者為女孩的2倍;高位畸形伴泌尿系畸形者占60%而低位畸形僅占20%
合併泌尿系異常的發生率與肛門直腸畸形類型有關,肛門直腸畸形的位置越高,合併泌尿系異常的可能性越大,且畸形嚴重。寺島報導在102例肛門直腸畸形中約2/3高位1/2中位和1/3低位合併泌尿系異常。
在合併泌尿系異常中,以膀胱輸尿管反流、腎積水和腎缺如多見。在宮野的病例中19%(24例)為膀胱輸尿管反流,在高、中位肛門直腸畸形中占30%~40%,其中一半為重度返流低位僅占8%。
Stephens報導兩組病例,在30例直腸畸形中有13例伴發泌尿系畸形28例肛門畸形中9例有泌尿系畸形,而兩組中約有半數未做泌尿系檢查,可見伴發泌尿系畸形者很多
生殖系畸形也是肛門直腸畸形常見的伴發畸形,因為它們有共同的胚胎髮生學基礎,即均為午菲管發育異常所致。在男嬰常見尿道下裂、隱睪等,Cook報導在253例男嬰肛門直腸畸形中有14例伴尿道下裂在收集的1272例病兒中有39例合併尿道下裂另外也有伴發少見的陰莖前陰囊畸形者。
在高位肛門直腸畸形的女嬰中,內生殖器畸形也較常見,包括陰道缺如、雙陰道、陰道閉鎖子宮陰道積液子宮缺如、雙角子宮等。據Hasse統計,在1272例肛門直腸畸形中有10例陰道缺如。
近年來中國醫科大學附屬醫院無選擇地對28例肛門直腸畸形病兒進行了靜脈腎盂造影(IVP)等泌尿系檢查,屬高位肛門直腸畸形的15例,中位10例低位3例伴有各種瘺道者占71.4%,其中膀胱瘺1例,尿道瘺11例,泄殖腔瘺3例陰道瘺2例舟狀窩瘺1例,會陰瘺2例;無瘺8例。發現伴有上尿路異常者10例(35.7%),其中右腎及輸尿管缺如2例,左腎及輸尿管缺如4例,右重腎及重輸尿管1例,右腎發育不良1例雙側腎異位1例,膀胱輸尿管返流1例,輸尿管開口異位1例。共7種異常11個病變(其中1例同時患有雙側病變)。本組病例中以腎及輸尿管缺如最多,占6例發生在高位肛門直腸畸形者5例發生在中位肛門直腸畸形者1例
伴有泌尿系瘺以外的下尿路異常3例(10.7%),膀胱外翻、尿道上裂、膀胱憩室及尿道憩室各1例,共4種異常(其中1例同時伴有膀胱外翻和尿道上裂)。伴發其他各種畸形者17例(60.7%),其中有2種以上其他畸形者7例(25%),多見骨骼和生殖系異常、先天性心臟病、神經系統及消化道異常。在骨骼異常中以腰骶椎異常多見,有9例(32.1%),半椎體、腰椎融合各3例,脊柱裂4例,骶骨發育不良及骶骨平直各1例;其次為多指、肋骨和骨盆多發畸形。生殖系異常可見隱睪、雙陰道及隱匿陰莖等先天性心臟病2例均為室間隔缺損。還可見直腸重複畸形及大腦發育不全、會陰部脂肪瘤和腰骶部脊膜膨出等畸形。
肛門直腸畸形合併有泌尿系異常早期多無泌尿系症狀,往往易被忽視。因不能及時診斷和處理,使很多泌尿系畸形不能得到矯治。一些重要的泌尿系異常如腎積水膀胱輸尿管反流等,由於未能及時診斷和治療,易造成尿路感染,甚至導致腎功能障礙。因此對肛門直腸畸形,特別是高、中位畸形患兒常規進行泌尿系檢查十分必要。一旦確診合併重症腎積水或膀胱輸尿管反流應及早矯治。
脊椎特別是腰骶椎畸形也是肛門直腸畸形經常伴發的畸形,自Hohl於1852年首次報導肛門直腸畸形病兒伴發骶椎畸形以來,有關報導逐漸增多但伴發腰骶椎畸形的發生率各作者報導不一,為2.5%~66%。腰骶椎畸形的發生率與肛門直腸畸形類型有關。
Stephens報導30例高位肛門直腸畸形,其中17例合併骶椎異常;而28例低位畸形中僅有2例合併骶椎異常。里村報導26例高位畸形中11例(41%)合併腰骶椎畸形,在25例低位畸形中7例(28%)合併腰骶椎畸形有人報導,2/3的高位畸形兒(男、女均同)和1/3的男性低位畸形兒伴發骶骨畸形,而女性低位畸形兒則很少有骶椎異常。
李正等對113例肛門直腸畸形病兒(其中高位32例,中位40例低位41例)腰骶椎正、側位X線片進行研究,結果發現:X線片上腰骶椎完整者97例其中有異常者52例(53.6%),其發生率明顯高於對照組(9.5%)高中低位畸形骶椎異常的發生率分別為66.6%、58.3%和40.5%,其中多發性異常則各為50%、25%和0.97例腰椎X線片上,11例(11.3%)有腰椎異常,即腰椎骶化5例,半椎體4例,腰椎融合發育不全和鏇轉畸形各1例,共12例次。
在113例骶椎X線片上,57例(50.4%)有各種異常,共94例次。骶椎異常的發生率與畸形類型有關,高位畸形最多為62.5%;中位55%;低位為36.6%。23例(20.4%)有2個以上多發性異常,最多者有3~4個在高中、低位畸形中多發性骶椎異常的發生率依次為40.6%、22.5%和2.4%。在骶椎異常中,骶椎缺如和發育不全最多共41例次(36.2%),在X線片上有各種不同的表現,可以是全骶椎缺如,也可以是部分缺如,即有1個或2個以上椎體缺如;有的表現為椎體側塊缺如多為一側,也有兩側者;有的椎體發育較小,或同時有2~3個椎體融合在一起。其次骶骨平直或反曲即骶骨正常彎曲消失,甚至遠位骶骨向後方反曲,本組共30(26.5%)例次。隱性骶椎裂也較常見共28(24.8%)例次多發生在第1、2骶椎,個別的為全骶椎裂。有9例(8.0%)為骶椎腰化。
有些腰骶椎異常無臨床症狀,對患兒無任何影響,如一部分隱性腰骶椎裂、腰椎骶化和骶椎腰化等。正常人有隱性脊椎裂者為5%~6%。本組有28例肛門直腸畸形患兒有此改變占24.8%,明顯高於正常人腰椎半椎體和鏇轉畸形可造成脊柱側彎,隨年齡增長而加重。骶椎缺如或發育不全與肛提肌和骶神經的發育有密切關係。有人報導,第4、5骶椎缺如者肛提肌發育正常;第3骶椎以下缺如,該肌發育薄弱;而第12骶椎以下缺如該肌不發育。一般骶椎缺如與骶神經缺如是一致的,第3骶椎以上缺如或發育不全可嚴重地累及肛提肌及其支配神經,導致術後肛門功能障礙,引起完全的或部分的大小便失禁。但也有少數病例,骶骨雖缺如骶神經尚存在其功能良好。有人報導的246例中,51例有骶骨畸形,其中12例有嚴重的功能障礙在Ленюшкин報導的無肛畸形術後大小便失禁的106例患兒中,約10%(12例)的患兒伴有骶椎全部缺如或骶椎裂。可見嚴重的骶椎異常可影響預後。未見全骶椎缺如者,第3骶椎以下缺如或發育不全者18例,其中8例(44.4%)術後排便功能有明顯障礙,無論是臨床評分和(或)直腸肛管測壓、肛門外括約肌肌電圖和鋇灌腸X線檢查評分均較差,可能與骶骨異常有關
李正等觀察到伴骶骨平直或反曲者共30例(26.5%),其發生率與肛門畸形類型有關。骶骨平直或反曲與排便功能的關係尚不清楚。但其中16例單純骶骨平直或反曲無其他改變的患兒中,有5例(31.3%)經臨床和客觀檢查有排便功能障礙,值得進一步研究。
鑒於肛門直腸畸形伴發脊椎畸形的發生率很高,因此,對每個肛門直腸畸形病兒,特別是高、中位畸形者應做脊椎X線片檢查,以便及早了解伴發畸形,對估計預後和及時採取治療措施有益。
除脊柱畸形外,肛門直腸畸形伴發四肢骨骼畸形者也常有報導。有人報導肛門畸形病兒中有2%有橈骨發育不全,在有下肢畸形的病兒中更為常見。
肛門直腸畸形伴發心臟及大血管畸形者也較常見。在一組384例肛門直腸畸形中,有心臟和大血管畸形者39例占10.2%。其中219例高、中位畸形中有30例心臟大血管畸形,165例低位畸形中有9例法洛四聯症和巨大室間隔缺損是最常遇到的畸形,其死亡率高,一般須急診做心外科手術。近來有人指出凡肛門直腸畸形手術經過順利,而術後吸奶無力、氣急及皮膚蒼白者,經檢查多伴有室間隔或房間隔缺損。
肛門直腸畸形還可伴發各種各樣的消化道其他畸形,據Acgill報導約占10%,如該畸形伴食管閉鎖的報導越來越多。在利物浦醫院384例肛門畸形病兒中31例合併食管閉鎖,占8.1%。伴發巨結腸的發生率說法不一,有人報導88例肛門直腸畸形中2例合併先天性巨結腸,占2.3%;而Kiesewetter等人收集35個醫療中心的病例,發現其發生率為3.4%;Santulli等人調查結果在1166例肛門畸形中僅1例伴無神經節細胞症。我們近30年來治療肛門直腸畸形1000餘例,也只見到1例伴發巨結腸。肛門直腸畸形也可伴發腸閉鎖、環狀胰腺、腸重複畸形、腸鏇轉不良等畸形。因此,對肛門直腸畸形病兒腹部X線片上腹腔無氣體者,應警惕消化道其他部位也有梗阻另外肛門直腸畸形也可合併有罕見的多種畸形組合在一起的複雜畸形,如內臟外翻、膀胱小腸裂等。總之,肛門直腸畸形病兒同時可伴發其他臟器畸形,而且可以有幾種畸形同時存在。有的伴發畸形可直接影響預後,甚至危及病兒生命。因此對肛門直腸畸形病兒應進行全面檢查,特別是對泌尿系和骶椎檢查不容忽視,以免遺漏伴發畸形。
臨床表現
 先天性肛門直腸畸形
先天性肛門直腸畸形先天性肛門直腸畸形的種類很多,其臨床症狀不一,出現症狀時間也不同有的患兒生後即出現急性腸梗阻症狀有的生後很久才出現排便困難甚至少數患兒長期沒有症狀或症狀輕微。絕大多數肛門直腸畸形患兒,在正常肛門位置沒有肛門。嬰兒出生後24h不排胎便就應想到肛門直腸畸形,應及時進行檢查約有3/4的病例,包括全部無瘺的肛門直腸閉鎖和一部分雖有瘺但瘺口狹小不能排出胎糞或僅能排出少量胎糞者,如直腸膀胱瘺、尿道瘺等,餵奶後就出現嘔吐,吐出物為奶,並含有膽汁以後可吐糞樣物,腹部逐漸膨脹,病情日趨嚴重,如未確診和治療,6~7天即可死亡。另一部分病例,包括肛門直腸狹窄和有陰道瘺、前庭瘺及會陰瘺且瘺管較粗者,在生後一段時間內不出現腸梗阻症狀,而在數周、數月,甚至數年後出現排便困難、便條變細腹部膨脹,有時在下腹部可觸到巨大糞塊。此時已有繼發性巨結腸改變。
1.高位或肛提肌上畸形約占肛門直腸畸形的40%,男孩較女孩多見。不論是男孩或女孩往往有瘺管存在,但因瘺管較細,幾乎都有腸梗阻症狀。此種患兒在正常肛門位置皮膚稍凹陷色澤較深但無肛門。患兒哭鬧或用勁時,凹陷處不向外膨出,用手指觸摸該處也沒有衝擊感。
女孩往往伴有陰道瘺,多開口於陰道後壁穹隆部。這類患兒外生殖器發育不良,呈幼稚型。因無括約肌控制,糞便經常從瘺口流出,易引起生殖道感染以後便秘越來越重,逐漸形成繼發性巨結腸,表現為腹部膨隆,常常可以觸到巨大糞塊患兒全身情況不佳,有慢性中毒症狀。
泌尿系瘺幾乎都見於男孩,女孩罕見。從尿道口排氣和胎糞是直腸泌尿系瘺的主要症狀。膀胱瘺時因胎糞進入膀胱與尿混合,患兒在排尿的全過程中尿呈綠色,尿的最後部分色更深,同時可排出瀦留在膀胱內的氣體。如壓迫膀胱則胎糞和氣體排出更多。在不排尿時,因受膀胱括約肌控制,無氣體排出。直腸尿道瘺時僅在排尿開始時排出少量胎糞,不與尿相混而以後的尿液則是透明的。因為沒有括約肌控制,從外尿道口排氣與排尿動作無關。
上述症狀對診斷泌尿系瘺有重要意義,但由於瘺管的粗細不同,或往往被黏稠的胎糞所堵塞,出現的程度不一樣,甚至完全不出現。因此常規檢查患兒尿中有無胎糞成分是很必要的一次尿檢查陰性,不能除外泌尿系瘺的存在,必須多次檢查
有些病例可根據X線片膀胱內有氣體或液平面而確診有人指出,腸腔內有鈣化影也是診斷直腸泌尿系瘺的根據尿道膀胱造影時,造影劑往往僅能充滿瘺口部出現憩室樣陰影,而進入直腸內的造影劑很少。位於尿道膜部的瘺管較粗時,導尿管沿尿道後壁插入可通過瘺口進入直腸。
伴有泌尿系瘺的病例在新生兒期如未得到矯治可反覆發生尿道炎、陰莖頭炎和上尿路感染,甚至出現外瘺另外,這些患兒合併脊椎畸形者較為常見,骶神經的發育也受累,其分支支配膀胱和肛門括約肌即或在行畸形矯治手術之後,也可能有尿便失禁現象
2.中間位畸形約占15%,這類畸形過去被一些人歸入高位畸形而另一些人則將此歸入低位畸形。無瘺者直腸盲端在尿道球海綿肌邊緣或陰道下端附近,恥骨直腸肌包繞直腸遠端。有瘺者其瘺管開口於尿道球部、陰道下段或前庭部。其肛門部位的外觀與高位畸形相似也可自尿道或陰道排便。探針可通過瘺道進入直腸,用手指觸摸肛門部可觸到探針的頂端。
女孩直腸前庭瘺較陰道瘺多見。瘺孔開口於陰道前庭舟狀窩部,也稱舟狀窩瘺瘺孔較大,嬰兒早期通過瘺孔能維持正常排便,甚至較大兒童也能正常排便,僅在稀便時有失禁現象。如直腸前庭瘺的瘺口很窄其臨床表現與開口於外陰部的各種低位畸形相似然而通過瘺口插入探針,則探針向頭側走行而非向背側。嬰兒期因經常有糞便流出,如護理不周,在陰道前庭部經常有糞便,可引起陰道炎或上行性感染。
肛門直腸狹窄為罕見畸形,狹窄累及肛門及直腸下段,可能與肛門狹窄混淆瘺管造影可確定診斷。
3.低位或經肛提肌畸形約占肛門直腸畸形的40%。直腸末端位置較低在恥尾線以下。此種畸形多合併有瘺道,但較少並發其他畸形
臨床表現有的在正常肛門位置有凹陷,肛管被一層隔膜完全閉塞。隔膜有時很薄,呈深藍色。患兒哭鬧時隔膜明顯向外膨出,用手指觸摸時有明顯衝擊感,對刺激有明顯收縮。有的肛膜雖破,但不完全,其口徑僅有2~3mm,排便困難。有的肛門正常,但位置靠前,在正常肛門與陰囊根部或陰唇後聯合之間,稱會陰前肛門,一般臨床上無何症狀,不需治療。
一些男性低位肛門閉鎖患兒同時伴有肛門皮膚瘺管,其中充滿胎糞,而呈深藍色。瘺管開口於會陰部或更前一些至陰囊縫線或陰莖腹側的任何部位在女孩隱匿的胎糞不易看到,但如自瘺口插入探針,則緊挨皮下直接向後走行
在女孩中一些低位畸形靠近陰唇後聯合處的外陰部有一開口,其外觀與正常肛門相似,稱前庭肛門或外陰部肛門。在肛門前庭瘺,腸管已通過恥骨直腸肌,肛管末端通過一小瘺管與前庭相通。在臨床上此種瘺管與直腸前庭瘺所不同者,為插入瘺管口的探針稍向背側走而非頭側,用手指觸摸正常肛門處易觸到探針頭。
另外,還有一些罕見畸形,如女孩的會陰裂隙,在肛門與陰道前庭之間有一濕潤的具有上皮的裂隙。女嬰還有少見的泄殖腔畸形,其外陰發育呈幼稚型,大陰唇瘦小,僅見一個開口,尿便自此口排出。
併發症:
1.泌尿系感染高位或肛提肌上畸形伴有泌尿系瘺時,病兒可反覆發生尿道炎、陰莖炎和上尿路感染。
2.尿便失禁高位或肛提肌上畸形合併脊椎畸形者,若骶神經發育受累,因其分支支配膀胱和肛門括約肌肉故可有尿便失禁。
3.陰道炎直腸前庭瘺女病兒,因糞便經常自陰道前庭瘺流出,可引起病兒陰道炎或上行性感染。
診斷
先天性肛門直腸畸形的診斷在臨床上一般並不困難,但更重要的是準確的測定直腸閉鎖的高度,直腸末端與恥骨直腸肌的關係和有無泌尿系瘺以及脊柱畸形的存在,以便更合理的採取治療措施為此,應根據臨床症狀、體徵進行必要的輔助檢查。
檢查
 先天性肛門直腸畸形
先天性肛門直腸畸形1.X線檢查1930年Wangensteen和Rice設計了倒置攝片法診斷肛門直腸畸形,至今仍被廣泛採用。其操作步驟是在生後12h以上先將病兒臥於頭低位5~10min,用手輕柔按摩腹部使氣體充分進入直腸。在會陰部相當於正常肛穴位置的皮膚上固定一金屬標記,或塗少量鋇劑做標誌,再提起病兒雙腿倒置1~2min,X線中心與膠片垂直,X線管球與患兒間距2米,雙髖併攏屈曲位(70°~90°)射入點為恥骨聯合在病兒吸氣時曝光。做側位和前後位攝片。盆腔氣體陰影與金屬標記間的距離即代表直腸末端的高度。在側位片上,從恥骨中點向骶尾骨交界處連一線,為恥尾線(PC線),再於坐骨嵴與恥尾線劃平行線為Ⅰ線盆腔氣體影高於PC線者為高位畸形,恰位於PC線與Ⅰ線之間者為中間位畸形,低於Ⅰ線者為低位畸形這對決定治療措施、選擇術式有重要意義。
王常林通過對50具新生兒盆腔屍體經10%福馬林液固定後解剖觀察恥骨直腸肌上、下緣最低點作與PC線的平行線分別為SS’和LL’,提出在X線照片上,以恥骨直腸肌上下緣最低點做為高、中、低位畸形的分界線較為合適。觀察結果,SS’線及LL’線分別距肛門(1.75±0.3)cm和(1.04±0.27)cm。為了便於臨床使用,故提出直腸盲端氣體陰影距肛穴1.8cm和1.1cm兩個距離標準,即盲端距肛穴1.8cm以上者為高位,1.1cm以下者為低位,在1.1~1.8cm之間者為中間位畸形此標準與PC線、M線I線的位置關係基本一致同時套用該法判定肛門直腸畸形病兒45例41例判定正確,直腸盲端所處位置和恥骨直腸肌的解剖與本法術前判定相符合;4例判定失誤,主要原因是投照條件不當,其中1例為早產兒合併肺炎、硬腫症,24h攝片時,結腸氣體很少,導致判斷失誤。
值得注意的是,倒立側位X線片有時遇到下列情況可造成誤差:①檢查過早(生後12h以內者),腸道氣體尚未充盈達到直腸末端;②檢查時病兒倒置時間少於1~2min;③X線射入角度不合適及在病兒呼氣時曝光。
在觀察X線倒置位平片時,同時觀察骶尾骨有無畸形、反曲、融合、半椎體及缺如等改變同時應觀察膀胱內有氣體或液平面,或在腸腔內有鈣化的胎便影,直腸盲端呈鳥嘴狀改變等是診斷泌尿系瘺的可靠方法。發現此種改變可行逆行性尿道膀胱造影,此時可見造影劑充滿瘺道或進入直腸對確定診斷有重要價值對有結腸造瘺的病兒採用經腸腔或瘺道造影,可以了解瘺管長度、瘺道走行方向及直腸末端的水平等。
2.B超檢查套用超音波斷層掃瞄器,機械扇形掃描探頭頻率為3~5MHz患兒檢查無需特殊準備,取平臥截石位,探頭接觸病兒肛穴處會陰皮膚,作矢狀切面掃查可獲得肛門直腸區聲像圖會陰部皮膚呈細線狀強回聲;骶骨椎體常顯示串珠狀排列強回聲伴有後方聲影,第1骶椎較寬,並向前下方傾斜,構成骶曲起始部,易辨認;骶椎前方一般可見直徑約1cm的管狀結構回聲,管腔內多為無回聲區,其中間可有氣泡的強回聲;直腸前上方可見充盈的膀胱,膀胱壁呈細線狀強回聲,內部則為無回聲區,而膀胱後方可見回聲增強。
檢查時如發現會陰皮膚的回聲顯示不清可在皮膚表面加水囊以增加水囊與皮膚界面的清晰度。如果有會陰瘺、前庭瘺者可經瘺管外口插入導管注入生理鹽水20~40ml,最好取頭高足低30°位使直腸盲端充分充盈,防止水外溢。按上述方法掃描,不但可以顯示直腸盲端與肛門皮膚間距而且可以觀察瘺管走向和長度。直腸膀胱瘺者膀胱內均見遊動的強回聲或較強回聲光點,按壓下腹部時光點明顯增多。
正常直腸在骶前穿過盆隔肛提肌與外括約肌至肛門通往體外。直腸閉鎖形成的直腸盲端與會陰皮膚之間常有軟組織相隔。盆腔底的軟組織在超聲檢查中常顯示非均質強回聲,直腸盲端多充滿胎糞,在超聲檢查中則顯示盲管形低回聲。但要注意盲腔內胎糞稀稠可導致回聲有差異,稀薄胎糞呈低回聲或近似無回聲,如盲腔內含有氣體則有強回聲區,體位改變時強回聲位置常隨之移動。患兒哭鬧腹內壓有改變時,管腔盲端即隨呼吸上下擺動此時應待直腸盲端圖像移至與皮膚最近位置時停幀,或改變探頭方向使呈冠狀切面掃查而停幀測量與肛區皮膚最短距離。
全學模套用B超檢查肛門直腸畸形15例除1例手術發現與B超測值相差1.0cm外,其餘14例誤差均在0.3cm以內。此外汪楚文也報導套用B超檢測肛門閉鎖合併直腸瘺16例其中合併前庭瘺8例,會陰瘺5例,膀胱瘺及尿道瘺3例。直腸盲端與肛周皮膚間距測量與外科檢測結果符合15例,僅1例誤差>5mm。
3.CT檢查肛門括約肌群包括內、外括約肌及恥骨直腸肌,其形成及發育程度是決定肛門直腸畸形患兒預後最重要的因素。套用CT直接了解直腸盲端與恥骨直腸肌環的關係,對提高嬰幼兒肛門直腸畸形的治療效果是極重要的。
套用CT儀進行盆腔掃描,檢查前病兒禁食4~6h用硫賁妥鈉10~15mg/kg,
或氯胺酮3~6mg/kg肌內注射。仰臥位,雙下肢伸直固定於檢查台上,以恥骨聯合下緣為零點,每5mm為一平面,從-5mm位開始倍精掃描,依次向頭端斷層檢查,共獲得8個斷面圖像。也有人自會陰部皮膚表面向頭側端與體軸成直角,每隔5mm作一次掃描,CT水平窗中心為正30,寬250恥骨聯合下緣作為標準掃描平面在掃描限幅內,用電子計算機算出骨盆出口肌肉分布面積。骨盆出口三角是以兩坐骨結節下緣為底邊恥骨聯合為頂點,用計算機求出三角形面積用骨盆出口肌肉分布的面積與骨盆出口三角面積之比乘以100%,即算出二者之比。正常兒隨年齡增長面積比值基本相同為37.2%±5.7%。不同類型肛門直腸畸形患兒比值有明顯差異,低位與中位肛門畸形患兒肛周肌肉分布及面積比值基本接近正常,而高位畸形肌肉分布範圍小,比值低。因此,套用CT提供可靠的形態學資料,有助於手術方案的設計。郎詩民等報導2例無肛CT檢測情況:1例2天男嬰高位肛門直腸閉鎖,CT檢查:在閉孔肌平面清楚顯示腸管通過肌環;在-5mm平面中線肛門處可見括約肌軟組織團塊影另一例亦為男性,6個月因高位肛門直腸閉鎖行結腸造瘺術後CT檢查:在閉孔內肌平面無腸腔影,僅在恥骨後方有一包括後尿道和肌環的軟組織團塊影;在肛門區平面僅顯示一線狀形、無括約肌團塊影。術後10天複查,在腸腔內插入14號導尿管,可見腸管外有較厚肌層環繞;術後3個月複查患兒控制排便能力良好。
4.MRI檢查檢查前0.5h口服5%水合氯醛溶液1.5ml/kg患兒仰臥位,在正常肛穴位置和瘺孔處用魚肝油丸做標誌可對盆腔做矢狀、冠狀和橫斷面掃描,每5mm一個斷面,矢狀、冠狀斷面從直腸中央向外和向後掃描,橫斷面從肛門標誌處向上掃描
正常新生兒肛周肌群在MRI各斷面上表現為:恥骨直腸肌在矢狀面上位於PC線部位骶尾骨前方;冠狀面位於直腸遠端兩側;橫斷面位於直腸遠端前後方。肛門外括約肌在橫斷面位於直腸遠端,呈圓形肌束圍繞於肛門周圍;在矢狀、冠狀面位於肛管前後或左右。
我們對9例肛門直腸畸形病兒行MRI檢查,年齡1~3天6例,5~6個月3例高位畸形4例,中間位畸形2例低位畸形3例,結果顯示:中位畸形1例,高位畸形3例伴有骶骨平直和(或)反曲,第3、4、5骶椎融合和第5骶椎缺如各1例。5例低、中位畸形恥骨直腸肌發育良好而4例高位畸形中,僅1例該肌發育較好2例發育薄弱(均經手術證實),另1例不清。肛門外括約肌發育正常者5例,其中1例高位畸形在直腸盲端下方尿道後方有豐富的肌組織影像說明外括約肌發育良好。1例中位和3例高位畸形中外括約肌發育較差,並在直腸盲端與肛穴間充滿大量脂肪組織,其中3例已手術者均被證實。
無瘺的肛門直腸畸形患兒直腸盲端擴張,充滿胎糞,可清晰地看到它與PC線的關係和距肛穴的距離。有瘺或結腸造瘺後的患兒直腸內空虛,在MRI片上可看到骶尾骨發育情況。在MRI片上不能顯示內括約肌改變,因為直腸盲端內充滿胎糞,腸壁菲薄而造瘺者直腸盲端空虛,腸壁影像不清。
對肛門直腸畸形行MRI檢查,可以觀察肛門周圍肌群的改變,同時可以判斷畸形類型和骶尾椎有無畸形。MRI對患兒無損害可從3個方面觀察肛周肌群的改變,較CT只做橫斷面更全面。為了獲得清晰影像,檢查前給予一定量鎮靜劑,肛穴處作好標誌,必要時經瘺口注入氣體充盈直腸盲端,使影像更清晰是非常必要的。
5.其他檢查有人套用穿刺的方法確定直腸末端的高度,即用消毒的注射器接粗針頭,從相當於正常肛門位置的中心向後上刺入邊向上推進邊抽吸,當針尖進入直腸末端時即有胎便或氣體排出,針刺入的長度即代表直腸末端與皮膚的距離。吸出胎便後,也可注入造影劑進行攝片以明確畸形的類型和腸道的位置此法對非常高的直腸閉鎖可能無效,且有危險,故應慎重
用探針檢查瘺道也是明確瘺道的走行長度和寬度的簡便方法。如同時用指尖在正常肛門位置觸摸探針的頂端,可以粗略的估計盲端與皮膚的距離。
對有直腸陰道瘺的女孩,可用鼻鏡直接觀察瘺口的位置。
治療
 先天性肛門直腸畸形
先天性肛門直腸畸形1.按患病類型來分先天性肛門直腸畸形的治療方法根據其類型及末端的高度不同而異。
(1)會陰前肛門無狹窄排便功能無障礙者:一般不需治療。肛門或直腸下端輕度狹窄一般採用擴張術多能恢復正常功能。擴張方法是用特製的金屬探子,自肛門插入直腸內,最初1次/d留置15~20min,逐漸改為隔天1次或每周1~2次一般持續6個月左右,直到排便正常,且能保持狹窄不再復發為止。探子應由小到大,直到能通過食指為止。並應教會家長用手指進行擴肛。如肛門顯著狹窄,須行手術治療。
(2)低位肛門直腸畸形:包括有瘺和無瘺者,以及肛門閉鎖伴前庭瘺者應行會陰肛門成形術。對無瘺或有瘺但不能維持排便者,一般需在生後1~2天內完成手術。對伴有較大瘺孔如前庭瘺肛門狹窄等,生後在一段時間內尚能維持正常排便,可於6個月左右施行手術。
會陰肛門成形術的方法是於正常肛門位置做“X”形切口,各長1.5cm,切開皮膚及皮下組織用止血鉗向深部鈍性分離找到直腸盲端,此時透過腸壁,可見深色胎糞。用組織鉗鉗住直腸盲端,或用0號絲線於直腸盲端縫合2針支持線,縫線僅穿過漿肌層,不要穿透腸壁全層,以免胎糞自針孔處外溢。用止血鉗鉗夾小紗布球,緊貼腸壁進行鈍性分離,先游離直腸後壁及兩側壁,最後游離直腸前壁。前壁距尿道(或陰道)很近,為了防止損傷尿道(或陰道)於該處注入0.25%利多卡因溶液1~2ml,使腸壁與尿道(或陰道)壁分開,即可較易分離。游離直腸要充分一般以使直腸盲端自然突出於皮膚切口之外0.5~1.0cm為宜。
用4號絲線於直腸壁前、後、左右行漿肌層縫合4針,固定於括約肌按“+”字形切開直腸盲端,排出胎糞。將皮膚切口的4個皮瓣尖插入直腸盲端“+”字形切口的間隙中用1號絲線將直腸準確地與皮膚縫合先在四角皮瓣的八個尖端縫合然後在兩縫線間再縫合1~2針保留一條縫線以固定肛管。選適當粗的肛管包以凡士林紗布,插入直腸內4~5cm。
肛門會陰瘺者,其直腸盲端與肛門皮膚的距離較近,多在1cm以內。於手術開始前,自瘺孔填入凡士林紗條以防術中糞便外流沿瘺孔兩側及後緣呈半環形切開皮膚並於其中點向後方延長1.5cm。游離直腸後壁及兩側壁,前壁不需游離。待腸壁充分游離後,剪去已游離的部分瘺孔邊緣,並沿瘺管縱行切開直腸後壁1~1.5cm將直腸壁與括約肌縫合固定3針,結節縫合直腸與皮膚。
對較少見的陰囊或陰莖皮膚瘺,手術時不必游離和切除瘺管,僅於其基底部即對入直腸的部分切斷結紮即可。此瘺管以後多發生機化而閉鎖。如瘺管不能閉鎖於2~3歲時將其切除。
肛門前庭瘺者,在肛門正常位置作“X”形切口以保存陰唇後聯合的完整性切開皮膚、皮下組織,向深部做鈍性分離,以顯露直腸盲端及瘺管。游離直腸後壁及兩側壁,於近前庭處先橫斷瘺管,再自下而上地將直腸前壁與陰道後壁分開(如先游離瘺管,再將其切斷,因接近瘺管的直腸與陰道後壁緊密黏著,如勉強進行分離,則易造成陰道或直腸損傷)然後將遠端瘺管由前庭處的瘺孔向外翻出並於靠近瘺管口處將其貫穿縫合結紮。縫合直腸與皮膚。
(3)中位肛門直腸畸形:常伴直腸尿道球部瘺或低位直腸陰道瘺等。因瘺管位置特殊從盆腔或會陰部均不易暴露,應行骶會陰肛門成形術。此手術宜在患兒6個月左右施行故對無瘺和伴直腸尿道瘺的中位畸形患兒,應先作橫結腸造瘺,以解除梗阻症狀。伴低位直腸陰道瘺者,其瘺孔較大,在一段時間內尚能維持正常排便,則不必作結腸造瘺。
骶會陰肛門成形術是於尾骨尖下方作半弧形切口,長約5cm,沿正中線切開肛尾肌膜靠近中線向深部分離以免損傷支配肛提肌的神經中位畸形恥骨直腸肌包繞於瘺管及直腸盲端的後下方,用直角鉗緊貼直腸做鈍性分離,邊分離邊向前推進,張開兩鉗葉直至鉗尖插入肌環。動作要輕柔,以免撕斷肌纖維。
在肛門處作“X”形切口,於外括約肌中插入止血鉗,並輕柔地向上分離,使之與自骶部切口插入的直角鉗相遇。然後將一條膠皮帶穿過外括約肌中心及恥骨直腸肌環從兩切口引出作牽引用,用宮頸擴張器逐漸將兩肌環擴大至能通過直腸為止。
對伴有尿道或陰道瘺者,應在直視下游離瘺管並將其切斷、縫扎或縫合殘端。充分游離直腸,使直腸無張力地能自然下降到肛門切口為止。從肛門切口插入組織鉗夾住直腸盲端,將其緩慢地牽至肛門。直腸與皮膚用絲線縫合。伴尿道瘺者應作恥骨上膀胱造瘺,並取出尿道內導尿管。
(4)高位肛門直腸畸形:包括無瘺和有瘺以及直腸閉鎖的病例。確定診斷後,為了挽救患兒的生命,應作橫結腸或乙狀結腸造瘺術,以解除梗阻症狀。待6個月後,再行骶腹會陰肛門成形術。
在尾骨尖下方橫行切開皮膚2~3cm,沿中線切開肛尾肌膜並向深部分離。高位畸形時恥骨直腸肌向前上方移位,位於尿道或陰道壁後方顯露該肌後,用直角鉗緊貼尿道或陰道後壁,邊張開兩鉗葉進行分離,邊向前推進,直至鉗尖插入肌環。然後將直角鉗尖端向後至會陰部新肛門處,作會陰部切口,使之與骶部切口相通,並將一膠皮帶穿過外括約肌中心及恥骨直腸肌環自兩切口引出。
開腹游離直腸,沿直腸周圍向下作鈍性分離,顯露直腸盲端。如有瘺管,應將其充分顯露並鉗夾、切斷斷端用碘酊、酒精處理後絲線作貫穿縫合結紮同時應剝除殘端遺留的黏膜,以免分泌的黏液積聚。充分游離直腸、乙狀結腸,使其能無張力地達到會陰切口之外。用組織鉗通過會陰部切口進入腹腔,鉗住直腸並向下牽引直腸盲端至會陰部切口之外。在牽引時,防止發生扭轉抽出膠皮帶,結節縫合直腸與皮膚形成肛門。
骶腹會陰黏膜下切除肛門成形術:骶及會陰部手術步驟與Stephens骶會陰肛門成形術相同。腹部手術,在盆腔腹膜返折處切開盆底腹膜,游離乙狀結腸。在腹膜返折部位的直腸漿肌層與黏膜間注入生理鹽水使黏膜與肌層分離環行切開漿肌層,保持黏膜完整。沿黏膜下層用銳性及鈍性向遠端分離直至直腸盲端後,結紮、切斷瘺管,並切開肌鞘下端。然後用一把大彎血管鉗通過會陰切口穿過肛門外括約肌中心、恥骨直腸肌及直腸盲端切口,鉗夾直腸近端,自會陰部切口拖出形成肛門。此手術避免了盆腔剝離面廣,損傷大的缺點,但有形成直腸肌鞘內積液、積膿以及尿道憩室的可能。
2.手術方法
(1)後矢狀入路肛門直腸成形術:1980年由Vires和Pena提出的,適宜於高、中位肛門直腸畸形。
自骶尾關節上方到肛穴前方正中線上用針形電刀切開皮膚、皮下組織。在電刺激下觀察兩側肌肉的發育情況,並從正中將橫紋肌複合體分為左、右2部分,顯露直腸盲端。先游離直腸後壁及兩側壁最後游離直腸前壁。如有尿道(陰道)瘺,於直腸盲端縫支持線,切開腸腔,直腸前壁中央凹陷處即為瘺口。在直視下距瘺口3mm處切開腸壁一圈,用4-O~6-O尼龍無損傷針線縫合閉鎖瘺口,並自下而上游離直腸前壁直到直腸在無張力的情況下達到肛門處為止。如果直腸達不到肛門處或有張力,可將直腸周圍纖維膜牽拉到緊張處,作多個不同水平的小橫切口使之松解,可延長直腸約3~5cm,或開腹游離直腸。如直腸盲端極度擴張,難以通過肌肉複合體時,應將直腸後壁作倒“V”形剪裁,使其直徑為1.2~1.5cm直腸置於左右2部分橫紋肌複合體之間,將肌肉複合體與腸壁縫合固定數針,縫合修復肌肉複合體及外括約肌。直腸與肛周皮膚縫合形成肛門。
本手術的優點是所有操作都在直視下進行術野清晰,避免了盲目地切開分離,將手術損傷減少到最小程度儘量保留直腸及肛周組織,恢復直腸與其周圍組織的正常解剖關係,以便術後獲得較好的肛門控制功能。
(2)泄殖腔畸形修復術:對泄殖腔畸形應於出生後立即作結腸造瘺,使糞流改道,保持泄殖腔出口清潔防止發生尿路感染根治手術的時間應根據患兒情況、畸形複雜程度及術者的經驗而定一般以6個月以後手術為宜。也有人主張陰道成形術應在青春前期完成。
術前應從泄殖腔開口做逆行造影,以了解畸形類型是常見型、高位型或低位型不但要了解泄殖腔的大小、尿道瘺和直腸瘺的高度,還要了解子宮的發育情況和有無畸形,以便選擇術式。
 先天性肛門直腸畸形
先天性肛門直腸畸形手術取後正中矢狀切口。從骶骨中段到泄殖腔外口處,在電刺激引導下,在中線上分開外括約肌和肛提肌。充分游離泄殖腔管,顯露直腸進入泄殖腔的入口。在該處直腸黏膜縫數根牽引線,於直腸和陰道共壁之間做黏膜下分離。一般分離到距陰道開口以上2cm直腸與陰道壁開始獨立分開分離直腸的長度直至能無張力地達到肛門皮膚為止。直腸分開後可顯露陰道後壁。用同樣的方法將陰道從尿道與陰道的共壁間做黏膜下分離。此處分離比較困難,因為陰道從後麵包繞尿道一半以上,而且組織彈性差。陰道分離後往往出現陰道前壁缺血。陰道分離得越長,缺血越嚴重,越易出現尿道陰道瘺。陰道游離充分後修復尿道,特別是共同管兩側橫紋肌對控制排尿有重要作用。圍繞著事先置入膀胱的尿管修復尿道,縫合兩層,然後將陰道在尿道後方縫合於皮膚上對分離時嚴重損傷陰道前壁的病例,為防止出現尿道陰道瘺,應將陰道扭轉90°,即使有血循環的陰道側壁接觸尿道縫線。如陰道不能達到會陰皮膚,可選用下列方法作陰道成形:
①皮膚陰道成形術:適用於陰道缺損較短的病例。皮瓣從未來陰道部位的兩側皮膚或陰唇皮膚形成,應為保留皮下組織具有良好血液供應的全厚皮瓣,兩側皮膚缺損縫合閉合。
②腸管陰道成形術:陰道缺損較多或無陰道的病例採用帶腸系膜的迴腸或乙狀結腸修復陰道。即在尿道修復和直腸游離之後,開腹並切取一段帶腸系膜的腸管,自會陰拖出腸管近端縫合於子宮或陰道下緣,腸管遠端縫合在會陰部皮膚上
最後作直腸修復形成肛門,即將直腸置於肛提肌與外括約肌中心,並將肌肉與腸壁縫合固定數針,同時重建會陰體。
泄殖腔畸形修復術均需做恥骨上膀胱造瘺術。術後2周傷口癒合後,應擴張肛門及陰道。新陰道不能隨身體發育而成比例的擴大,因此,陰道擴張要持續到青春期。
預後:
肛門直腸畸形的治療效果近年來已有明顯改善總病死率由過去的25%~30%降至近年的10%左右手術死亡率已降到2%左右。
目前仍有約1/3的病例術後有不同程度的肛門功能障礙。有些患兒功能障礙嚴重給患兒及其家屬造成長期的,甚至是終身的痛苦和煩惱。肛門直腸畸形的位置越高術後排便功能障礙的發生率越高,程度越嚴重。術後排便障礙的發生率,功能障礙高位畸形者為86.4%中位畸形為47.9%,低位畸形為27%。
關於較客觀準確地判定肛門排便功能,李正、王練英等綜合國內外有關資料及自己的經驗,並考慮到設備條件,提出肛門成形術後肛門功能綜合評定標準。推薦以便意、失禁的有無及其程度判定的臨床評分與以直腸肛門測壓肌電圖和鋇灌腸X線檢查等指標判定的客觀評分法。在眾多客觀檢查指標中,選出以代表肛門內括約肌功能為主的肛管高壓區長度,代表肛門外括約肌功能的肌電圖靜止時波幅,代表恥骨直腸肌位置和功能的肛管直腸角,以及代表盆底橫紋肌收縮功能的直腸肛管收縮壓差為指標。該法較其他方法更能全面準確地反映排便功能。對排便功能差的患兒,經系統的生物反饋肛門功能訓練仍無改善者,應行括約肌成形術進行矯治。
