發病機制
病因:賴利-戴綜合徵的病因至今尚不完全清楚近年來研究發現本病為常染色體隱性遺傳性周圍性神經病所有的患者均有兩個複製基因缺陷致病基因位於9號染色體短臂31~33區上確切病因不明。系常染色體隱性遺傳,具有家族性,本徵發病可能與兒茶酚胺代謝異常有關,由於多巴胺β羥化酶(DβH)活力降低,使多巴胺轉變為去甲腎上腺素過程發生障礙。患者體內兒茶酚胺特別是去甲腎上腺素含量減低,故對外源性去甲腎上腺素反應敏感,而且對胰島素造成低血糖狀態的自身恢復較慢。但若投予腎上腺素,便能迅速恢復。其次不僅發現患者對乙醯甲膽鹼反應敏感,且許多膽鹼能神經功能不全症狀可迅速改善,並發現患者角膜、虹膜和淚腺的乙醯膽鹼轉換酶不足,表明乙醯膽鹼代謝異常對發病亦起重要作用。
發病機制:本病為自主神經系統先天性異常,患兒感覺神經節交感神經節及副交感神經節中健康搜尋的神經元明顯減少這些神經元在患者一生中不斷喪失。交感神經末梢數量減少健康搜尋是病人血液循環中去甲腎上腺素多巴胺、β-羥化酶較少的原因。由於外周組織交感神經支配減少使腎上腺能受體過敏以致腎上腺髓質釋放兒茶酚胺可引起過度應激反應雖有些症狀提示累及中樞神經系統,但中樞神經系統未見類似病變智力屬正常範圍。
也有研究指出健康搜尋患兒尿中的去甲腎上腺素、腎上腺素代謝產物香草醯扁桃酸(VMA)降低,高香草酸(HVA)大量增多,也可能是由於患兒體內兒茶酚胺代謝異常,去甲腎上腺素及其衍生物形成障礙所致。
本病的病理改變主要是頸交感神經節發育不良神經元數目和體積均減少。主要表現在丘腦背內側核、頸髓與胸髓側角灰質細胞、背根神經節及交感神經節健康搜尋的異常改變,節前神經元減少,腦幹網狀結構變性蝶齶神經節、睫狀神經節的神經細胞異常;此外脊髓背柱背根脊丘束等有脫髓鞘改變少數發現脊髓交感神經節有色素變性。
病理:中樞神經系統無恆定的異常,有的可見顯著的丘腦囊腫,有的發現橋腦和延髓網狀結構退變,有的還見到脊髓側索神經纖維減少或後索改變,以後周圍神經發育不良。腸神經叢可見胞漿內粒狀空泡形成。
臨床診斷
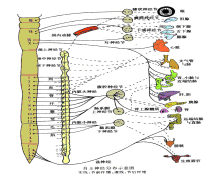 賴利-戴綜合徵
賴利-戴綜合徵1.本病多自出生後即感覺喪失和交感神經功能障礙,智慧型正常或低下,進展緩慢。患兒無性別差異,嬰兒期生長發育緩慢常伴發作性嘔吐、腹瀉或便秘肌痙攣、運動功能障礙、共濟失調Charcot病理性關節及口腔潰瘍等
2.患兒出生時體重低於正常嬰兒哭聲小而短促,吸吮力弱,吞咽功能差,易患吸人性肺炎,哭時淚水極少或無淚是本病的主要特徵。檢查可見肌張力低,腱反射減低或消失瞳孔對光調節異常,角膜反射消失,結膜乾燥無淚液,舌尖光滑,舌蕈狀乳頭缺失有廣泛性痛、溫覺的輕度減退,對疼痛刺激無反應健康搜尋,可見皮膚紅斑肢體發紺發涼異常多汗流涎或缺乏唾液、味覺障礙,以及癇性發作等。
3.在幼兒期可出現自主神經危象表現為情緒不穩,易激惹自閉行為減少、體溫易變心率及呼吸頻率不穩定血管運動障礙是本病的特徵之一,如血壓波動不穩,常出現直立性低血壓及發作性高血壓。
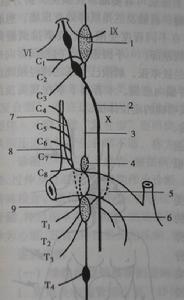 賴利-戴綜合徵
賴利-戴綜合徵5.患兒常因肺炎夭折,其他死亡原因為睡眠猝死和心跳驟停。對病人進行細緻周到的照顧和護理可生存到30~40歲病人也能生育正常的嬰兒。
併發症:可有原因不明的高熱,或並發吸入性肺炎健康搜尋。情緒常不穩定偶發嘔吐脫水及胃腸道症狀等。約40%的病人可有抽搐發作。
診斷:本病的診斷主要根據家族史,嬰幼兒期發病患兒哭時無淚舌蕈狀乳頭缺失自主神經症狀多變,尿中高香草酸(homovanillicacid,HVA)顯著增加用1:1000磷酸組胺(histaminephosphate)前臂皮內注射患兒無痛感局部無紅暈區和偽足用0.0625%毛果芸香鹼(pilocarpine)滴眼5min滴1次,共滴4次可見患兒眼瞼下垂。
鑑別診斷:本症應與下列兩種疾病相鑑別
1.急性全自主神經病急性起病,臨床表現為視力模糊瞳孔對光及調節反應異常出汗少,無淚液直立性低血壓,尿瀦留等多數病例在數周或數月後逐漸自發恢復。
2.Sjögren綜合徵主要特徵為流淚、唾液多汗腺及胃液的分泌都缺陷,常伴有角結膜炎鼻炎咽炎腮腺水腫及周圍性的腦神經麻痹等。
檢查治療
 賴利-戴綜合徵
賴利-戴綜合徵實驗室檢查:
1、血及腦脊液檢查無特異性。
2、基因檢測可發現基因缺陷。
其它輔助檢查:
1、患兒皮內注射組胺常無疼痛及紅暈反應
2、用2.5%醋甲膽鹼(乙醯甲膽鹼)毛果芸香鹼液滴眼能引起小瞳症,而對去甲腎上腺素液瞳孔反應卻正常,可協助診斷
3、腦電圖常顯示異常;腦幹聽覺誘發電位也示異常。
相關檢查:腦脊液
根據上述植物性神經功能紊亂的症狀及體徵,結合實驗室檢查可診斷。腦電圖、骨關節X線檢查等可能有助診斷。
治療
本病尚無特殊的藥物治療方法以對症治療為主吞咽困難的患者可給予鼻飼,肺部感染可適當套用抗生素多汗、流涎可服用阿托品類藥物,流淚障礙及角膜潰瘍可給對症處理,各種維生素鎮靜劑如地西泮氯丙嗪,以及抗癲癇藥物等已證實有一定的療效明顯的脊柱畸形可以考慮進行手術矯形。主要為預防感染、縫瞼術,麻醉有高度危險,預後差。
預防措施
預後:因肺炎嘔吐發作、脫水、癲癇或小兒尿毒症肺水腫等併發症多在兒童期死亡;若早期診斷,及時預防合併症及處理,不少患者可以生存至成年期。
預防:遺傳病治療困難,療效不滿意,預防顯得更為重要。預防措施包括避免近親結婚推行遺傳諮詢攜帶者基因檢測及產前診斷和選擇性人工流產等,防止患兒出生。
