
概述
脆性X染色體綜合徵是一種X連鎖智力低下疾病。在X染色體長臂末端有一脆性部位(即細絲部位),且經G帶證實。其臨床表現為智力低下,以男性患者顯著,並有語言障礙及行為異常,表現語言遲緩、單調、多言、孤獨、膽小。男性患者有巨睪特徵,少數患者在青春期前即有大睪丸出現,可伴有過度伸直的指關節、大手、大足及皮紋異常。產前檢查羊水可助早期診斷,必要時進行人工流產。
病因學
 脆性X染色體綜合徵
脆性X染色體綜合徵致病的FMR-1(Fragile-Xmentalretardation)基因在1991年被證明,由於其CGG重複過度擴增,導致不正常的甲基化,因此,無法正常產生FMR-1基因所編碼的蛋白(FMRP)。
正常情況下,FMR1基因很穩定的由親代傳給子代。此症患者的FMR1基因出現三核甘酸CGG重複擴增次數異常。當FMR-1基因CGG數低於54時,會穩定地遺傳給子代;在55-200之間,則稱之為準突變(pre-mutation),其本身並無症狀,但由女性傳遞給子女時,可能會進一步擴增CGG,達到全突變(full-mutation),即CGG大於200,甚至數千次之多,而導致子女罹患此症。
發生率
根據國外的調查,約1/350女性是此症的攜帶者,攜帶此缺陷的婦女若生下男嬰,患此症的機率高達50%。男性中的發生率約1/1,000-/1,250,在女性為1/2,000-1/2,500。此症有明顯的家族好發現象,後代成員患病的比例會愈高。
遺傳模式
染色體
 脆性X染色體綜合徵
脆性X染色體綜合徵此症的遺傳模式比較特殊。一般正常女性染色體為XX,男性則為XY,若1個X染色體異常,女性可以在另一個正常X染色體保護下變成隱性,而男性會因為缺乏保護而顯現症狀,這也就是為何女性攜帶率高,症狀卻更容易反應在男性身上。
攜帶有準突變基因的孕婦,可檢測胎兒的FMR-1基因,防止生出患病的後代。
家族中若有智障或發展遲緩、早期停經、情緒障礙的女性,則一定要進行基因檢查。
造成男性容易發病的另一個原因是:隱性攜帶的母親在遺傳給男孩時,會有一段不穩定的基因CGG序列突然擴增,造成患者在情緒、行為、學習及語言上出現障礙,進入青春期的男性患者,80%還會有睪丸變大的現象。
致病基因
現今在X脆性部們已發現了致病基因FMR-1,它含有(CGG)n三核甘酸重複序列,後者在正常人約為30拷貝,而在正常男性傳遞者和女性攜帶者增多到150~500bp,稱為小插入,相鄰的Cpg島未被甲基化,這種前突變(premutation)無或只有輕微症狀。女性攜帶者的CGG區不穩定,在向受累後代傳遞過程中擴增,以致在男性患者和脆性部位高表達的女性達到1000~3000bp,相鄰的CpG島也被甲基化。這種全突變(fullmutation)可關閉相鄰基因的表達,從而出現臨床症狀。由前突變轉化為完全突變只發生母親向後代傳遞過程中。根據對脆性部位DNA序列的了解,現已可用RFLP連鎖分析、DNA雜交分析、PCR擴增等方法來檢出致病基因。
常見模型
1、DNA複製過程中新生的DNA鏈和模板鏈在重複區域發生錯誤配對兒
〓〓〓〓〓〓〓〓〓
↓
〓〓〓~~~~~~
2、DNA雙鏈斷裂的重組修復有關。
3、DNA單鏈空缺修復有關。
4、其他原因。
另外需要說明的是脆性X染色體綜合徵只是近年來發現的眾多與三核甘酸擴增有關的人類遺傳病之中的一種。在人類所含有的可能十種三核甘酸重複序列中,其擴增可以導致人類遺傳病的還有GAA/CTT,CTG/CAG等。
臨床表征
幼兒時期 脆性X染色體綜合徵
脆性X染色體綜合徵女孩子的症狀通常較少,程度也較輕。可能會有的症狀包括:關節過度伸展及頭部跟臉部的特徵,在女孩子有時候這些生理特徵可以單獨發生。
生理特徵
臉部長而窄、大頭、柔軟而突出的大耳朵、突出的下顎、寬闊的鼻樑延伸至鼻尖、高而拱起的上顎。也可能有眼睛虹膜層呈淡藍色、眼眥贅肉、眼瞼下垂、近視、遠視、斜視、眼球震顫。
出生後
有結締組織發育不良的現象,所以可能造成全身肌肉張力較低以及關節過度伸展、關節較不穩定、扁平足的現象。還可能有心臟方面的問題,如二尖瓣脫垂症。
青春期之後
通常會有巨睪,臉部較長與巨睪症會開始明顯。
其它症狀
疝氣、關節脫臼、關節異常,以及較少見的脊柱側彎以及手指前端較大。此症的男孩要特別提防重複發作的中耳炎,所以應特別注意中耳的狀況及聽力問題。大約有20%患兒可能會有癲癇,部份患兒會有腦部構造或生理結構異常,例如:腦室過大或小腦異常。約80%男性患兒與30%女性患兒有智慧型遲緩或不足的現象。另外還常伴隨有感覺統合異常及注意力不足症等障礙。
診斷檢查
對所有非特異性的性連鎖MR均應考慮為本徵。臨床表現中以語言障礙為特徵的MR,長臉、大耳、巨睪和精神行為異常是脆性X染色體綜合徵的診斷要點。
染色體分析示脆點表達率≥4%即為陽性。染色體檢查通常在低或無葉酸和胸腺嘧啶的培養基中進行培養,但是陽性率低,不能測出脆性X染色體綜合徵攜帶者。
一般的內分泌檢查無特殊發現。EEG可見含有θ、α和β頻率的低幅複合波,間有短陣發放性4—7c/s的θ節律。葉酸治療有效則有助於診斷。
基因診斷是日前脆性X染色體綜合徵的診斷可靠的方法,包括DNA連鎖分析、Southern印跡雜交法、PCR技術等。直接DNA連鎖分析費時;Southern印跡雜交有較高的靈敏性及特異性,可以同時檢測CGG的拷貝數和甲基化狀態,PCR技術靈敏度高,通常作為篩查診斷。
治療
患者可以通過早期特殊療育計畫得到改善,根據專科醫師的經驗,患者年紀愈小,愈早接受早期療育的治療,治療效果也愈好。部分嚴重的行為問題,可使用藥物來治療。
實驗研究
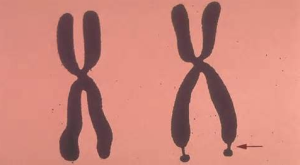 單一核苷酸的改變就可引發脆性X染色體綜合徵
單一核苷酸的改變就可引發脆性X染色體綜合徵研究者表示,正常的胚胎幹細胞在SNP位點及活性複製區域上具有一個胸腺嘧啶鹼基,而脆性X染色體細胞則有一個胞嘧啶鹼基,在對攜帶脆性X前突變的子代胚胎幹細胞進行研究後,研究人員發現,這些細胞有胸腺嘧啶鹼基及正常的複製模式,也就是說隨著時間延續其不會出現重複核苷酸序列的擴張。
這項研究顯示,當FMR1基因從母體遺傳給後代後,以胞嘧啶來替代胸腺嘧啶或許就會使得DNA複製區域失活,從而增加DNA重複擴張的風險,進而引發個體患脆性X染色體綜合徵。
