成份
本品主要成份為索磷布韋,其化學名稱為:(S)-異丙基2-((S)-(((2R,3R,4R,5R)-5-(2,4-二氧-3,4-二氫嘧啶-1(2H)-基)-4-氟-3-羥基-4-甲基四氫呋喃-2-基)甲氧基)-(苯氧基)磷基胺基)丙酸酯
化學結構式:
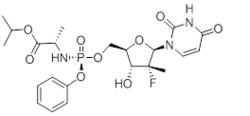 索磷布韋片
索磷布韋片分子式:CHFNOP
分子量:529.45
性狀
本品為薄膜衣片,除去包衣後顯類白色。
適應症
本品適於與其他藥品聯合使用,治療成人與 12 至 < 18 歲青少年的慢性C型肝炎病毒 (HCV) 感染。
關於 HCV 基因型比活性,請參見【注意事項】和【藥理毒理】。
規格
400mg
用法用量
本品的治療應由在慢性 HCV 感染患者管理方面有豐富經驗的醫生實施並監測。
劑量
成人
推薦劑量為每日一次,每次口服一片 400 mg 片劑,隨食物服用。
本品應與其他藥品合用。不推薦單藥治療。另請參閱與本品合用藥品的處方信息。本品聯合治療的推薦合用藥品和治療持續時間見表 1。
表 1:Sovaldi 聯合治療成人推薦的同服藥品和治療持續時間
 索磷布韋片
索磷布韋片* 包括合併感染人類免疫缺陷病毒 (HIV) 的患者。
a. 對於先前接受過治療的 HCV 基因型 1 感染患者,尚無海外 3 期數據。對於接受 Sovaldi、利巴韋林和聚乙二醇干擾素 α 聯合治療的中國受試者,已獲得相關數據(請參見【注意事項】和【臨床研究】,在存在慢性基因型 1、2、3 或 6 HCV 感染的中國成年患者中的臨床療效和安全性”章節)。
b. 應考慮到治療持續時間有可能延長而超過 12 周,最長達 24 周;尤其是對於那些具有一種或多種曾與基於干擾素的治療應答率較低相關的因素(例如,晚期纖維化/肝硬化、基線病毒濃度高、黑色人種、IL28B 非 CC 基因型以及先前對聚乙二醇干擾素 α 和利巴韋林治療無反應)的亞組。
c.請參閱下面的特殊患者人群-等待肝移植的患者。
與 Sovaldi 合用時,利巴韋林的劑量需基於體重(< 75 kg = 1000 mg,≥ 75 kg = 1200 mg),且需將此劑量分成兩次隨食物口服。
有關與針對 HCV 的其他直接作用抗病毒藥物合用的信息,請參見【注意事項】一節。
成人劑量調整
不建議減少 Sovaldi 的劑量。
如果索磷布韋與聚乙二醇干擾素 α 合用時,患者出現可能與該藥物有關的嚴重不良反應,那么應減少聚乙二醇干擾素 α 劑量或停止使用。有關如何減少和/或停止聚乙二醇干擾素 α 給藥的其他信息,請參閱聚乙二醇干擾素 α 處方信息。
如果患者出現可能與利巴韋林有關的嚴重不良反應,則應調整利巴韋林劑量或停藥(如果合適),直至不良反應緩解或嚴重程度降低。表 2 提供了根據患者的血紅蛋白濃度和心功能狀態進行劑量調整和停藥的指導方針。
表 2:成人中與 Sovaldi 共同給藥時利巴韋林的劑量調整指導方針
 索磷布韋片
索磷布韋片在利巴韋林因實驗室檢測異常或臨床表現而被停用後,可以嘗試以每天 600 mg 的劑量重新開始給予利巴韋林,並進一步增加劑量至每天 800 mg。不過,不建議將利巴韋林增加到最初指定劑量(每天 1000 mg 至 1200 mg)。
兒童人群
在 12 至 < 18 歲的青少年中,Sovaldi 的推薦劑量為每日一次,每次口服一片400 mg 片劑,隨食物服用。
本品應與其他藥品合用。不推薦單藥治療。本品聯合治療的推薦治療方案和持續時間見表 3 和表 4。
表 3:接受 Sovaldi 治療的 12 至 < 18 歲青少年的推薦治療方案和持續時間
 索磷布韋片
索磷布韋片表 4:在 12 至 < 18 歲青少年中利巴韋林與 Sovaldi 聯合治療的給藥建議
 索磷布韋片
索磷布韋片*利巴韋林的日劑量基於體重而定,並且分兩次隨食物口服。
兒童人群中的劑量調整
不建議減少 Sovaldi 的劑量。
如果患者出現可能與利巴韋林有關的嚴重不良反應,則應調整利巴韋林劑量或停藥(如果合適),直至不良反應緩解或嚴重程度降低。關於利巴韋林劑量調整或停藥指南,請參閱利巴韋林處方信息。
成人和青少年的停藥
如果與 Sovaldi 合用的其他藥品被永久停用,那么也應停用 Sovaldi(請參見【注意事項】)。
特殊患者人群
老年人
對於老年患者,無需調整劑量(請參見【藥代動力學】)。
腎功能損害
對於輕度或中度腎功能損害患者,無需調整 Sovaldi 劑量。尚未在重度腎功能損害(腎小球濾過率估計值 【eGFR】< 30 mL/min/1.73 m2)患者或需要進行血液透析的終末期腎病 (ESRD) 患者中確定 Sovaldi 的安全性和適當劑量(請參見【藥代動力學】)。
肝功能損害
對於輕度、中度或重度肝功能損害(Child-Pugh-Turcotte 【CPT】分級為 A、B 或 C)患者,無需調整 Sovaldi 劑量(請參見【藥代動力學】)。尚未確定 Sovaldi 在失代償性肝硬化患者中的安全性和療效。
等待肝移植的患者
等待肝移植的患者應依據個人狀況評估用藥效益與風險以此指導 Sovaldi 的用藥療程。
肝移植受者
在肝移植受者中,推薦 Sovaldi 與利巴韋林的合用時間為 24 周。推薦利巴韋林的起始劑量為 400 mg,分兩次隨食物口服。如果利巴韋林起始劑量的耐受性良好,可將劑量逐步上調至最高每日 1000 1200 mg(對於體重 < 75 kg 的患者,劑量為 1000 mg;對於體重 ≥ 75 kg 的患者,劑量為 1200 mg)。如果利巴韋林起始劑量耐受性不良,應根據血紅蛋白水平臨床指征降低劑量。
兒童人群
尚未確定 Sovaldi 在兒童及 18 歲以下青少年患者中的安全性和療效。
給藥方法
薄膜衣片適用於口服。應指導患者將片劑整片吞下。鑒於活性成分味苦,因此不可將薄膜衣片咀嚼或碾碎服用。該片劑應隨食物服用。
應指示患者如果在服藥 2 小時內出現嘔吐,則應再服用一片。如果在服藥超過 2 小時後出現嘔吐,則無需補服。這些建議基於索磷布韋和 GS 331007 的吸收動力學(表明大部分劑量在給藥後 2 小時內吸收)而定。
如漏服一劑藥物但仍在正常服藥時間後 18 小時內,則應指示患者儘快服用該片劑,之後患者應在平常用藥時間進行下一次服藥。若已超過 18 小時,則應指示患者等至平常用藥時間時進行下一次給藥。應指示患者不可服用兩倍劑量。
不良反應
國外臨床研究中成人的安全性特徵總結
在索磷布韋合用利巴韋林或合用聚乙二醇干擾素 α 和利巴韋林的治療期間,最常報告的藥品不良反應與利巴韋林和聚乙二醇干擾素 α 治療期間的預期安全性特徵一致,預期藥品不良反應的頻率或嚴重程度未增加。
不良反應的評估基於五項國外3期臨床研究(包括對照研究和非對照研究)的匯總數據進行。
在接受安慰劑、12 周索磷布韋 + 利巴韋林、16 周索磷布韋 + 利巴韋林、24 周聚乙二醇干擾素 α + 利巴韋林和 12 周索磷布韋 + 聚乙二醇干擾素 α + 利巴韋林治療的受試者中,因不良反應而永久停止治療的受試者比例分別為 1.4%、0.5%、0%、11.1% 和 2.4%。
國外臨床研究中成人的不良反應總結表
主要對 Sovaldi 與利巴韋林的合用(並用或不並用聚乙二醇干擾素 α)進行了研究。在此情況下,未發現特定發生於索磷布韋的藥品不良反應。在接受索磷布韋 + 利巴韋林或索磷布韋 + 利巴韋林 + 聚乙二醇干擾素 α 治療的受試者中,最常發生的藥品不良反應為疲勞、頭痛、噁心與失眠。
在索磷布韋 + 利巴韋林或索磷布韋 + 聚乙二醇干擾素 α + 利巴韋林的聯合治療中,發現以下藥品不良反應(表 5)。下面依據身體系統器官分類及發生頻率列出了不良反應。頻率規定如下:非常常見 (≥ 1/10)、常見(≥ 1/100 到 < 1/10)、少見(≥ 1/1000 到 < 1/100)、罕見(≥ 1/10000 到 < 1/1000)或極罕見 (< 1/10000)。
表 5:國外臨床研究中索磷布韋 + 利巴韋林或索磷布韋 + 聚乙二醇干擾素 α + 利巴韋林聯合治療時報告的藥品不良反應
 索磷布韋片
索磷布韋片a. SOF = 索磷布韋;b. RBV = 利巴韋林;c. PEG = 聚乙二醇干擾素 α。
慢性 HCV 感染的中國成人患者中的安全性特徵總結
在感染 HCV 的中國受試者中,索磷布韋合用利巴韋林或合用聚乙二醇干擾素 α 和利巴韋林時的安全性特徵與國外 3 期臨床研究中觀察到的安全性特徵基本相似。
在感染 HCV 的中國受試者中,因不良事件而永久停止治療(無論因果關係如何)的受試者比例較低:在接受 12 周索磷布韋 +利巴韋林治療、24 周索磷布韋+利巴韋林治療和 12 周索磷布韋+聚乙二醇干擾素 α+利巴韋林治療的受試者中,這一比例分別為 1.6% (1/64)、0.5% (1/195) 和 0.8% (1/130)。
在索磷布韋合用利巴韋林或合用聚乙二醇干擾素 α 和利巴韋林的治療期間,最常報告的不良事件與聚乙二醇干擾素 α 和利巴韋林治療期間的預期安全性特徵大體一致。
在索磷布韋與利巴韋林的 12 周合用治療期間,最常見的治療相關不良事件如下:網織紅細胞計數增加(21.9%,14/64 名受試者)、貧血(10.9%,7/64 名受試者)和血紅蛋白降低(10.9%,7/64 名受試者)。
在索磷布韋與利巴韋林的 24 周合用治療期間,最常見的治療相關不良事件如下:網織紅細胞計數增加(16.4%,32/195 名受試者)、血膽紅素升高(16.9%,33/195 名受試者)和貧血(10.3%,20/195 名受試者)。
在索磷布韋與聚乙二醇干擾素 α 和利巴韋林的 12 周合用治療期間,最常見的治療相關不良事件如下:發熱(35.4%,46/130 名受試者)、血小板計數減少(26.9%,35/130 名受試者)、中性粒細胞計數減少(26.9%,35/130 名受試者)、白細胞計數減少(24.6%,32/130 名受試者)、白細胞減少(20.8%,27/130 名受試者)、貧血(18.5%,24/130 名受試者)、中性粒細胞減少(16.9%,22/130 名受試者)、肌痛(14.6%,19/130 名受試者)、疲勞(13.8%,18/130 名受試者)、無力(13.8%,18/130 名受試者)、血紅蛋白降低(13.8%,18/130 名受試者)、頭痛(13.1%,17/130 名受試者)、頭暈(10.0%,13/130 名受試者)和血小板減少(10.0%,13/130 名受試者)。
其他特殊人群
HIV/HCV 合併感染
在合併感染 HCV/HIV 的受試者中,索磷布韋 + 利巴韋林的安全性特徵與 3 期臨床研究中在接受索磷布韋 + 利巴韋林治療且只感染 HCV 的受試者中觀察到的安全性特徵類似。
等待肝移植的患者
在肝移植前的 HCV 感染成人受試者中,索磷布韋 + 利巴韋林的安全性特徵與 3 期臨床研究中在接受索磷布韋 + 利巴韋林治療的受試者中觀察到的安全性特徵類似。
肝移植受者
在存在慢性C型肝炎病毒感染的肝移植成人受者中,索磷布韋 + 利巴韋林的安全性特徵與 3 期臨床研究中在接受索磷布韋和利巴韋林治療的受試者中觀察到的安全性特徵類似。在研究 0126 中,治療期間血紅蛋白降低情況非常常見,32.5% 的受試者(13/40 名受試者)的血紅蛋白降至 < 10 g/dL,其中 1 名還降至< 8.5 g/dL。8名受試者 (20%) 接受依泊汀和/或血液製品治療。在 5 名受試者 (12.5%) 中因不良事件而停用、調整或中斷研究藥物。
兒童人群
Sovaldi 在 12 至 < 18 歲青少年中的安全性和療效基於一項 2 期開放標籤臨床試驗中接受 12 周(基因型 2 患者)和 24 周(基因型 3 患者)Sovaldi +利巴韋林治療的 50 名患者的數據而定。觀察到的不良反應與在 Sovaldi +利巴韋林成人臨床研究中觀察到的不良反應一致(見表 5)。
選定不良反應的說明
心律失常
當索磷布韋與另一種 DAA(包括達拉他韋、西美瑞韋和來迪派韋)及合用藥物胺碘酮和/或其他降低心率的藥物聯合使用時,觀測到出現嚴重心動過緩和心臟傳導阻滯情況(請參見【注意事項】和【藥物相互作用】)。
疑似不良反應的報告
藥品被批准上市後疑似不良反應的報告十分重要。如此可持續監測使用該藥品的效益/風險平衡。在中國,要求醫療保健專業人員通過國家報告系統報告任何疑似不良反應。
禁忌
對活性成分或以下所列任一賦形劑過敏:
片芯:甘露醇、微晶纖維素、交聯羧甲基纖維素鈉、膠體二氧化矽、硬脂酸鎂。
薄膜包衣:聚乙烯醇、二氧化鈦、聚乙二醇、滑石粉、氧化鐵黃。
與強效 P-gp 誘導劑合用
腸內強效 P-糖蛋白 (P-gp) 誘導劑類藥品(利福平、利福布丁、聖約翰草 【Hypericum perforatum】、卡馬西平、苯巴比妥和苯妥英)。聯合用藥會顯著降低索磷布韋血漿濃度,並可能導致 Sovaldi 失去療效(請參見【藥物相互作用】)。
注意事項
概述
不建議 Sovaldi 以單藥治療形式給藥,應與其他藥品合用來治療C型肝炎感染。如果與 Sovaldi 合用的其他藥品被永久停用,那么也應停用 Sovaldi(請參見【用法用量】)。在開始使用 Sovaldi 治療前,請查閱聯合處方藥品的處方信息。
重度心動過緩和心臟傳導阻滯
當索磷布韋與另一種直接作用抗病毒藥(DAA,包括達拉他韋、西美瑞韋和來迪派韋)及合用藥物胺碘酮(加或不加降低心率的其他藥物)聯合使用時,觀測到出現嚴重心動過緩和心臟傳導阻滯情況。尚未確定機制。
整個索磷布韋加DAA 的臨床開發過程中,限制胺碘酮的合用。上述情況可能會危及生命,因此僅在不耐受或禁用其他替代性抗心律失常治療的情況下,才能在接受 Sovaldi 和另一種 DAA 治療的患者中使用胺碘酮。對於還在服用 β 受體阻滯劑的患者或有潛在心臟並存病和/或晚期肝病的患者,在與胺碘酮聯合用藥時發生症狀性心動過緩的風險可能會增加。
如果認為有必要合用胺碘酮,建議在開始 Sovaldi 和另一種 DAA 治療時對患者進行嚴密監測。應在適當的臨床環境中對確定存在較高慢脈性心律失常風險的患者進行 48 小時的持續監測。
由於胺碘酮的半衰期較長,還應對在過去幾個月內停用胺碘酮並即將開始 Sovaldi 與另一種 DAA 聯合用藥的患者進行適當的監測。
另外,還應提醒所有接受 Sovaldi 和另一種 DAA 與胺碘酮聯合給藥(加或不加其他可降低心率的藥物)的患者注意心動過緩和心臟傳導阻滯的症狀,並應建議他們出現此類症狀立即就醫。
接受過治療的基因型 1、4、5 和 6 HCV 感染患者
尚未在任何國外 3 期研究內,對接受過治療的基因型 1、4、5 和 6 HCV 感染患者中進行Sovaldi研究。因此,尚未確定此人群的最佳治療持續時間(另請參見【用法用量】)。治療這類患者時,應考慮到索磷布韋、聚乙二醇干擾素 α 和利巴韋林的治療持續時間可能延長至超過 12 周,最長達 24 周;特別是對於那些具有一種或多種曾與含干擾素的治療應答率較低相關的因素(晚期纖維化/肝硬化、基線病毒濃度高、黑色人種、IL28B 非 CC 基因型)的亞組。
在 3b 期研究 GS-US-334-0115 中,對於接受過治療的中國基因型 1 或 6 HCV 感染患者,接受 12 周 Sovaldi、聚乙二醇干擾素和利巴韋林治療時的 SVR12 率為 90.4% (47/52)(請參見【臨床研究】)。
基因型 5 或 6 HCV 感染患者的治療
支持基因型 5 和 6 HCV 感染患者接受 Sovaldi 治療的臨床數據十分有限。
基因型 1、4、5 和 6 HCV 感染的無干擾素治療
尚未在國外 3 期研究中對基因型 1、4、5 和 6 HCV 感染患者使用不含干擾素的 Sovaldi 治療方案進行研究。尚未確定最佳方案和治療持續時間。這些治療方案應僅用於對干擾素治療不耐受或不適用的患者以及急需治療的患者。在 3b 期研究 GS-US-334-0115 中,接受 24 周 Sovaldi 和利巴韋林治療的中國基因型 1 或 6 HCV 感染患者的合計 SVR12 率為 95.7% (66/69)(請參見【臨床研究】)。
與其他針對 HCV 的直接作用抗病毒藥物的合用
Sovaldi 應僅在基於現有數據確認效益大於風險的情況下與其他直接作用抗病毒藥品合用。無數據支持 Sovaldi 與特拉匹韋或博賽潑維聯合用藥。不推薦此類聯合給藥(另請參見【藥物相互作用】)。
妊娠以及與利巴韋林合用
當 Sovaldi 與利巴韋林或聚乙二醇干擾素 α/利巴韋林合用時,育齡女性及其男性伴侶在治療期間以及治療後一段時間內必須按照利巴韋林處方信息中的建議採取有效的避孕措施。有關其他信息,請參閱利巴韋林的處方信息。
與中度 P-gp 誘導劑合用
腸內中度 P gp 誘導劑類藥品(如奧卡西平和莫達非尼)可能會降低索磷布韋血漿濃度,導致 Sovaldi 療效降低。使用 Sovaldi 時不推薦合用此類藥品(請參見【藥物相互作用】)。
腎功能損害
尚未評估 Sovaldi 在重度腎功能損害 (eGFR < 30 mL/min/1.73 m2) 或需要血液透析的 ESRD 受試者中的安全性。此外,尚未確定適當劑量。對於肌酐清除率 (CrCl) < 50 mL/min 的患者,在使用 Sovaldi 與利巴韋林或聚乙二醇干擾素 α/利巴韋林進行聯合治療時,也請參閱利巴韋林的處方信息(另請參見【藥代動力學】)。
HCV/HBV(B型肝炎病毒)合併感染
已有報導發現,在直接作用抗病毒藥治療期間或之後,B型肝炎病毒(HBV)再活化的病例,其中個別報導出現致命情況。 在開始治療前,應對所有患者進行HBV篩查。 HBV/HCV合併感染患者有HBV再活化的風險,因此應根據現行臨床指南進行監測和管理。
兒童人群
不建議< 12 歲的兒童患者使用 Sovaldi,因為尚未確定該藥物在這些人群中的安全性和療效。
孕婦及哺乳期婦女用藥
育齡女性/男性和女性避孕
當 Sovaldi 與利巴韋林或聚乙二醇干擾素 α/利巴韋林合用時,必須極其小心,以避免女性患者和男性患者的女性伴侶懷孕。經證實,暴露於利巴韋林的各種動物皆出現了明顯致畸性和/或胚胎影響(請參見【注意事項】)。育齡女性及其男性伴侶在治療期間以及治療後一段時間內必須按照利巴韋林處方信息中的建議採取有效的避孕措施。有關其他信息,請參閱利巴韋林的處方信息。
妊娠
尚無孕婦使用索磷布韋的數據或此類數據非常有限(不足 300 例妊娠結局)。
動物研究表明對生殖毒性無直接或間接有害影響。最高試驗劑量下,在大鼠和兔中未觀察到對胎仔發育有任何影響。不過,相對於推薦臨床劑量下的人體暴露量,尚無法充分估計大鼠中所達到的索磷布韋暴露邊界比。
作為一種預防措施,妊娠期間最好避免使用 Sovaldi。
不過,在利巴韋林與索磷布韋合用時,關於妊娠期間利巴韋林用藥的禁忌症同樣適用(另請參見利巴韋林處方信息)。
哺乳
尚不清楚索磷布韋及其代謝產物是否會分泌到人乳中。
所得的動物藥代動力學數據顯示代謝產物分泌到乳汁中。
不能排除對於新生兒或嬰兒的風險。因此,哺乳期間不應使用 Sovaldi。
生育力
尚無 Sovaldi 影響人類生育力的相關數據。動物研究未表明會對生育力產生有害影響。
兒童用藥
尚未確定 Sovaldi 在兒童及 18 歲以下青少年患者中的安全性和療效。
老年用藥
索磷布韋的國外臨床研究包括 65 名年齡為 65 歲及 65 歲以上的受試者。在各治療組間,65 歲以上受試者的應答率與較年輕受試者的應答率相似。
藥物相互作用
索磷布韋是一種核苷酸藥物前體。口服 Sovaldi 後,索磷布韋很快被吸收,並進行廣泛的肝臟和腸道首過代謝。由羧酸酯酶 1 等酶催化的細胞內藥物前體水解裂解以及由核苷酸激酶催化的連續磷酸化步驟會形成具有藥理學活性的尿苷核苷類似物三磷酸鹽。超過 90% 的藥物相關物質系統暴露來自主要的非活性循環代謝產物 GS 331007,後者經由與形成活性代謝產物平行和連續的代謝途徑而產生。母體索磷布韋約占藥物相關物質系統暴露量的 4%(請參見【藥代動力學】)。在臨床藥理學研究中,出於藥代動力學分析目的,對索磷布韋和 GS 331007 均進行了監測。
索磷布韋是藥物轉運體 P gp 和乳腺癌耐藥蛋白 (BCRP) 的底物,而 GS 331007 不是。
腸內強效 P gp 誘導劑類藥品(利福平、利福布丁、聖約翰草、卡馬西平、苯巴比妥和苯妥英)可能會顯著降低索磷布韋的血漿濃度,導致 Sovaldi 療效降低,因此在使用 Sovaldi 時應禁用此類藥品(請參見【禁忌症】)。腸內中度 P gp 誘導劑類藥品(如奧卡西平和莫達非尼)可能會降低索磷布韋血漿濃度,導致 Sovaldi 療效降低。使用 Sovaldi 時不推薦合用此類藥品(請參見【注意事項】)。Sovaldi 與可抑制 P gp 和/或 BCRP 的藥品合用,可能會增加索磷布韋的血漿濃度但不會增加 GS 331007 的血漿濃度,因此,Sovaldi 可與 P gp 和/或 BCRP 抑制劑合用。索磷布韋與 GS 331007 都不是 P gp 和 BCRP 的抑制劑,因此預計不會增加屬於此類轉運體底物的藥品的暴露量。
索磷布韋在細胞內的代謝活化途徑由通常具有低親和力和高活性的水解酶與核苷酸磷酸化途徑介導,這些途徑不太可能受到合用藥品的影響(請參見【藥代動力學】)。
接受維生素 K 拮抗劑治療的患者
由於在 Sovaldi 治療期間肝功能可能會有變化,因此建議對國際標準化比值 (INR) 進行密切監測。
其他相互作用
下文表 4 中總結了有關 Sovaldi 與可能合用藥品的藥物相互作用信息(其中最小二乘方幾何平均值 (GLSM) 比的 90% 置信區間 (CI) 標示如下,在預先確定的等效性區間之內“↔”,以上“↑”或以下“↓”)。此表並未包含全部內容。
表 6:Sovaldi 與其他藥品之間的相互作用
 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片 索磷布韋片
索磷布韋片NA = 未提供/不適用
a. 在有/無索磷布韋的情況下合用藥物的藥代動力學平均值比 (90% CI),以及在有/無合用藥物的情況下索磷布韋和 GS 331007 的平均值比。無影響 = 1.00
b. 所有相互作用研究均在健康志願者中進行
c. 基於歷史對照進行比較
d. 以 Atripla 給藥
e. 生物等效性範圍為 80% 125%
f. 等效性範圍為 70% 143%
藥物過量
所記錄的索磷布韋最高給藥劑量是對 59 名健康受試者給予的 1200 mg 索磷布韋單一超治療劑量。在該研究中,此劑量水平下未觀察到不良影響,且不良反應的頻率和嚴重程度與安慰劑和索磷布韋 400 mg 治療組所報告的相關情況相似。更高劑量產生的影響尚不明確。
Sovaldi 用藥過量無特定解毒劑。如果發生用藥過量,必須監測患者是否有中毒跡象。Sovaldi 用藥過量的治療需要採取基本的支持措施,包括監測生命體徵以及觀察患者的臨床狀態。血液透析可有效去除(提取率 53%)主要循環代謝產物 GS 331007。4 小時血液透析可去除 18% 的給藥劑量。
臨床試驗
國外研究中的臨床療效和安全性
在五項國外3 期研究對索磷布韋療效進行了評估,總計包括 1568 名存在慢性基因型 1 至 6 C型肝炎病毒感染的成人受試者。一項研究在未接受過治療的患有慢性基因型 1、4、5 或 6 C型肝炎病毒感染的受試者中開展,對他們進行索磷布韋、聚乙二醇干擾素 α 2a 及利巴韋林聯合治療;其他四項研究在患有慢性基因型 2 或 3 C型肝炎病毒感染的受試者中開展,對他們進行索磷布韋和利巴韋林聯合治療,這四項研究分別針對未接受過治療的受試者,不耐受、不適用或不願接受干擾素治療的受試者,先前接受過含干擾素的方案治療的受試者,以及所有受試者(無論既往治療史或是否能接受干擾素治療)。這些研究中的受試者患有代償性肝病,包括肝硬化。索磷布韋的給藥劑量為 400 mg,每日一次。利巴韋林劑量為基於體重每日 1000–1200 mg,分兩次給藥,聚乙二醇干擾素 α 2a 劑量(如適用)為每周180μg。每項研究的治療持續時間固定,不受受試者 HCV RNA 水平的引導(無應答引導規則)。
臨床研究期間使用 COBAS TaqMan HCV 檢測(2.0 版)(與 High Pure System 結合使用)測定了血漿 HCV RNA 值。此檢測法的定量下限 (LLOQ) 為 25 IU/mL。所有研究均以持續病毒學應答 (SVR) 為判定 HCV 治癒率的主要終點,其定義為在治療結束後 12 周時 HCV RNA 低於 LLOQ (SVR12)。
針對基因型 1、4、5 和 6 慢性C型肝炎病毒感染受試者的海外臨床研究
未接受過治療的成人受試者- NEUTRINO(研究 110)
NEUTRINO 是一項開放標籤、單組海外研究,在未接受過治療的基因型 1、4、5 或 6 HCV 感染受試者中評估了索磷布韋與聚乙二醇干擾素 α 2a 和利巴韋林的 12 周聯合治療。
接受治療的受試者 (n = 327) 中位年齡為 54 歲(範圍:19 至 70);64% 的受試者為男性;79% 為白人;17% 為黑人;14% 為西班牙裔或拉丁裔;平均身體質量指數為 29 kg/m2(範圍:18 至 56 kg/m2);78% 的受試者的基線 HCV RNA 水平大於 6 log10 IU/mL;17% 患有肝硬化;89% 感染基因型 1 HCV,11% 感染基因型 4、5 或 6 HCV。表 7 列出了索磷布韋 + 聚乙二醇干擾素 α + 利巴韋林治療組的應答率。
 索磷布韋片
索磷布韋片基線時具有 IL28B C/C 等位基因 【94/95 (99%)】與具有非 C/C(C/T 或 T/T)等位基因 【202/232 (87%)】的受試者的 SVR12 率均較高。
在 28 名感染基因型 4 HCV 的患者中,27 名實現了 SVR12。本研究中,一名基因型 5 HCV 感染受試者和所有 6 名基因型 6 HCV 感染受試者實現了 SVR12。
針對基因型 2 和 3 慢性C型肝炎病毒感染受試者的國外臨床研究
未接受過治療的成人- FISSION(研究 1231)
FISSION 是一項隨機分配、開放標籤、陽性對照研究,在感染基因型 2 或 3 HCV 感染且未接受過治療的受試者中評估了 12 周索磷布韋 + 利巴韋林的治療相較於 24 周聚乙二醇干擾素 α 2a + 利巴韋林的治療。索磷布韋 + 利巴韋林和聚乙二醇干擾素 α 2a + 利巴韋林治療組所用的利巴韋林劑量分別為 1000–1200 mg/天(基於體重)和 800 mg/天(與體重無關)。按 1:1 的比例對受試者進行隨機分配,並按肝硬化(有或無)、HCV 基因型(2 和 3)和基線 HCV RNA 水平(< 6 log10 IU/mL 和 ≥ 6 log10 IU/mL)對患者分層。基因型 2 或 3 HCV 感染受試者的納入比例約為 1:3。
接受治療的受試者 (n = 499) 中位年齡為 50 歲(範圍:19 至 77);66% 的受試者為男性;87% 為白人;3% 為黑人;14% 為西班牙裔或拉丁裔;平均身體質量指數為 28 kg/m2(範圍:17 至 52 kg/m2);57% 的受試者的基線 HCV RNA 水平大於 6 log10 IU/mL;20% 患有肝硬化;72% 感染基因型 3 HCV。表 9 列出了索磷布韋 + 利巴韋林和聚乙二醇干擾素 α + 利巴韋林治療組的應答率。
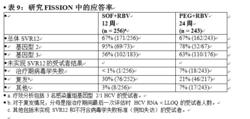 索磷布韋片
索磷布韋片索磷布韋 + 利巴韋林治療組與聚乙二醇干擾素 α + 利巴韋林治療組的總體 SVR12 率相差 0.3%(95% 置信區間:-7.5% 至 8.0%),此研究符合事先定義的非劣效性標準。
表 10 按 HCV 基因型列出了基線時患肝硬化的受試者的應答率。
 索磷布韋片
索磷布韋片針對干擾素不耐受、不適用或不願使用干擾素的成人的研究-POSITRON(研究 107)
POSITRON 是一項隨機分配、雙盲、安慰劑對照研究,在不耐受、不適用或不願接受干擾素治療的受試者中評估了 12 周索磷布韋 + 利巴韋林的治療 (n = 207) 相較於安慰劑的治療 (n = 71)。以 3:1 比例對受試者進行隨機分配,並按照肝硬化(有或無)進行分層。
接受治療的受試者 (n = 278) 中位年齡為 54 歲(範圍:21 至 75);54% 的受試者為男性;91% 為白人;5% 為黑人;11% 為西班牙裔或拉丁裔;平均身體質量指數為 28 kg/m2(範圍:18 至 53 kg/m2);70% 的受試者的基線 HCV RNA 水平大於 6 log10 IU/mL;16% 患有肝硬化;49% 感染基因型 3 HCV。不耐受、不適用或不願接受干擾素治療的受試者比例分別為 9%、44% 和 47%。大部分受試者先前未接受過 HCV 治療 (81.3%)。表 11 列出了索磷布韋 + 利巴韋林治療組和安慰劑治療組的應答率。
表 11:研究 POSITRON 中的應答率
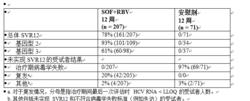 索磷布韋片
索磷布韋片與安慰劑組相比,索磷布韋 + 利巴韋林治療組的 SVR12 率具有統計學意義 (p < 0.001)。
表 12 按基因型列出了肝硬化和干擾素分類的亞組分析。
表 12:POSITRON 中按基因型列出的選定亞組的 SVR12 率
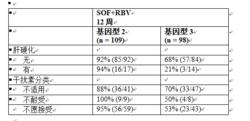 索磷布韋片
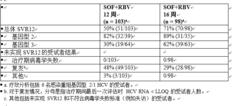
索磷布韋片針對先前接受過治療的成人的研究- FUSION(研究 108)
FUSION 是一項隨機、雙盲研究,在先前接受基於干擾素的治療未實現 SVR 的受試者(復發者和無應答者)中評估了 12 或 16 周索磷布韋 + 利巴韋林治療。以 1:1 比例對受試者進行隨機分配,並按照肝硬化(有或無)和 HCV 基因型(2 與 3)進行分層。
接受治療的受試者 (n = 201) 中位年齡為 56 歲(範圍:24 至 70);70% 的受試者為男性;87% 為白人;3% 為黑人;9% 為西班牙裔或拉丁裔;平均身體質量指數為 29 kg/m2(範圍:19 至 44 kg/m2);73% 的受試者的基線 HCV RNA 水平大於 6 log10 IU/mL;34% 患有肝硬化;63% 感染基因型 3 HCV;75% 為先前復發者。表 13 列出了 12 周和 16 周索磷布韋 + 利巴韋林治療組的應答率。
表 13:研究 FUSION 中的應答率
 索磷布韋片
索磷布韋片表 14 按基因型列出了針對肝硬化情況和先前 HCV 治療應答情況進行的亞組分析。
表 14:研究 FUSION 中按基因型列出的選定亞組的 SVR12 率
 索磷布韋片
索磷布韋片針對未接受過治療和先前接受過治療的成人的研究- VALENCE(研究 133)
VALENCE 是一項海外 3 期研究,在未接受過治療或先前接受基於干擾素的治療未實現 SVR 的受試者(包括患有代償性肝硬化的受試者)中評估了索磷布韋與基於體重使用的利巴韋林針對基因型 2 或 3 HCV 感染進行的聯合治療。將此研究設計為 12 周索磷布韋 + 利巴韋林與安慰劑的直接對比。然而,基於出現的數據,該研究被揭盲,所有基因型 2 HCV 感染受試者繼續接受 12 周的索磷布韋和利巴韋林,同時對基因型 3 HCV 感染受試者的治療延長至 24 周。在進行修正的時候,已經有十一名基因型 3 HCV 感染受試者完成了 12 周的索磷布韋和利巴韋林治療。
接受治療的受試者 (n = 419)中位年齡為 51 歲(範圍:19 至 74);60% 的受試者為男性;中位身體質量指數為 25 kg/m2(範圍:17 至 44 kg/m2);平均基線 HCV RNA 水平為 6.4 log10 IU/mL;21% 患有肝硬化;78% 感染基因型 3 HCV;65% 為先前復發者。表 15 列出了 12 周和 24 周索磷布韋 + 利巴韋林治療組的應答率。
表中未列出安慰劑組受者,因為他們均未實現 SVR12。
表 15:研究 VALENCE 中的應答率
 索磷布韋片
索磷布韋片表 16 按基因型列出了針對肝硬化情況和先前 HCV 治療暴露情況進行的亞組分析。
表 16:研究 VALENCE 中按基因型劃分的選定亞組的 SVR12 率
 索磷布韋片
索磷布韋片國外研究中 SVR12 與 SVR24 的一致性
在索磷布韋聯合利巴韋林或聯合利巴韋林和聚乙二醇干擾素治療後 SVR12 與 SVR24(治療結束後 24 周 SVR)是一致的,這表明陽性預測值和陰性預測值均為 99%。
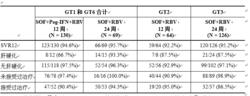
在存在慢性基因型 1、2、3 或 6 HCV 感染的中國成年患者中的臨床療效和安全性
在一項國內開放標籤臨床研究中對索磷布韋進行了研究,該研究評估了 12 周或 24 周索磷布韋和利巴韋林治療以及 12 周索磷布韋、聚乙二醇干擾素 α 2a 和利巴韋林聯合治療在未接受過治療或接受過治療的慢性基因型 1、2、3 或 6 C型肝炎病毒感染的中國受試者中的安全性和療效。
接受治療的中國受試者 (n = 389) 的平均年齡為 43 歲(範圍:19 至 79);49.9% 的受試者為男性且所有受試者 (100%) 均為中國受試者;平均身體質量指數為 23.2 kg/m2(範圍:16.2 至 35.9 kg/m2);總計 59/389 名受試者 (15.2%) 在基線時患有代償性肝硬化,162/389 名受試者 (41.6%) 有既往 HCV 治療經歷。在 162 名有既往 HCV 治療經歷的受試者中,既往治療失敗的原因為復發/治療期病毒學失敗(38.9%;63 名受試者)、無應答(21.0%;34 名受試者)和干擾素不耐受(40.1%;65 名受試者)。在 227 名未接受過治療的受試者中,大多數受試者(89.9%;204 名受試者)被視為干擾素適用。
大多數受試者攜帶 IL28B CC 等位基因 (76.9%)。基線 HCV RNA 總體平均值 (SD) 為 6.4 (0.71) log10 IU/mL,80.2% 的受試者的基線 HCV RNA 值 800,000 IU/mL;51%(199 名受試者)的受試者感染基因型 1 或 6 HCV、16%(64 名受試者)的受試者感染基因型 2 HCV 以及 32%(126 名受試者)的受試者感染基因型 3 HCV。表 17 按基因型和治療組列出了應答率。
表 17:研究 0115 中按基因型和治療組列出的中國受試者的 SVR12 率
 索磷布韋片
索磷布韋片HCV RNA 的分析使用了適於與 High Pure System 結合使用的 Roche TaqMan V 2.0 檢測,定量限值為 25 IU/mL。
SVR12 是停止研究治療後 12 周的持續病毒學應答 (HCV RNA < LLOQ)。
對於缺失的 SVR12 值,如果其前後值為被稱為成功的值(即,“< LLOQ TND”或“< LLOQ,檢測到”),則將其推定為成功,否則,將該缺失的 SVR12 值推定為失敗。TND = 未檢測到目標。
在所有治療組中,SVR12 率較高,範圍為 92.2% 至 95.7%。無論何種治療,除 1 人外其他基因型 6 HCV 感染受試者 (35/36) 均實現了 SVR12,94.5% 的基因型 1 HCV 感染受試者 (154/163) 實現了 SVR12。對於接受過治療的基因型 1 HCV 感染受試者,接受 12 周索磷布韋 +利巴韋林 +聚乙二醇干擾素 α 和 24 周索磷布韋+利巴韋林治療時的 SVR12 率分別為 90% (38/42) 和 94% (48/51)。在所有基因型中,21 名患者未實現 SVR12;20 名患者復發,1 名基因型 2 HCV 患者在完成治療之前死亡。
特殊人群中的臨床療效和安全性
合併感染 HCV/HIV 的患者- PHOTON 1(研究 123)
在一項開放標籤臨床研究中對索磷布韋進行了研究,該研究評估了 12 周或 24 周索磷布韋和利巴韋林治療在合併感染慢性基因型 1、2 或 3 C型肝炎病毒和 HIV 1 的受試者中的安全性和療效。感染基因型 2 和 3 病毒的受試者未接受過治療或接受過治療,而感染基因型 1 病毒的受試者之前未接受過治療。對於未接受過治療的基因型 2 或 3 HCV 感染的受試者,治療持續時間為 12 周;對於接受過治療的基因型 3 HCV 感染和基因型 1 HCV 感染的受試者,治療持續時間為 24 周。受試者接受 400 mg 的索磷布韋和基於體重的利巴韋林劑量(體重 < 75 kg,劑量為 1000 mg;體重 ≥ 75 kg,劑量為 1200 mg)。受試者未接受過抗逆轉錄病毒治療且 CD4+ 細胞計數大於 500 個細胞/mm3,或在病毒學上 HIV 1 受到抑制且 CD4+ 細胞計數大於 200 個細胞/mm3。進入研究時,95% 的患者接受過抗逆轉錄病毒治療。
表 18 按基因型和先前 HCV 治療暴露情況列出了應答率。
表 18:研究 PHOTON-1 中的應答率
 索磷布韋片
索磷布韋片表 19 按基因型列出了針對肝硬化情況的亞組分析。
表 19:研究 PHOTON 1 中按基因型劃分的選定亞組的 SVR12 率
 索磷布韋片
索磷布韋片等待肝移植的患者-研究 2025
在一項評估移植前給予索磷布韋與利巴韋林對預防移植後 HCV 再感染的安全性和療效的開放標籤臨床研究中,在接受肝移植前感染 HCV 的受試者中研究了索磷布韋。研究的主要終點為移植後病毒學應答(pTVR,移植後 12 周 HCV RNA < LLOQ)。
達到米蘭標準的肝細胞癌 (HCC) 受試者,無論感染哪種基因型的 HCV,每天均接受 400 mg 的索磷布韋和 1000–1200 mg 的利巴韋林,最長達 24 周,後來修正為 48 周或直至接受肝移植時(以先發生者為準)。在接受索磷布韋和利巴韋林的 61 名受試者中,大多數受試者感染基因型 1 HCV,44 名受試者為 CPT A 級,17 名受試者為 CPT B 級。在這 61 名受試者中,44 名受試者在接受長達 48 周的索磷布韋和利巴韋林治療後接受了肝移植;肝移植時,41 名受試者 HCV RNA < LLOQ。表 20 中描述了移植時 HCV RNA < LLOQ 的 41 名受試者的病毒學應答率。對於移植時 HCV RNA < LLOQ 的受試者,移植前的病毒抑制持續時間是 pTVR 的最佳預測因子。
表 20:肝移植時 HCV RNA < LLOQ 受試者的移植後病毒學應答率
 索磷布韋片
索磷布韋片在 24 周停止治療的患者中,根據方案,復發率為 11/15。
肝移植受者-研究 0126
在一項開放標籤臨床研究中對索磷布韋進行了研究,該研究評估了 24 周索磷布韋和利巴韋林治療在患有慢性C型肝炎病毒感染的肝移植受者中的安全性和療效。符合條件的受試者的年齡 ≥ 18 歲,且他們在篩選之前 6 至 150 個月時接受了肝移植。受試者在篩選時的 HCV RNA ≥ 104 IU/mL,且移植前已有記錄證明存在慢性 HCV 感染的跡象。利巴韋林的起始劑量為每日 400 mg,分次服用。如果受試者的血紅蛋白水平維持在 ≥ 12 g/dL,則在第 2 周、第 4 周及之後每 4 周增加利巴韋林劑量,直至達到基於體重而定的適當劑量(對於 < 75 kg 的受試者,每日劑量為 1000 mg;對於 ≥ 75 kg 的受試者,每日劑量為 1200 mg)。第 4 至 24 周期間的中位利巴韋林劑量為每日 600 mg–800 mg。
納入 40 名受試者(33 名感染基因型 1 HCV,6 名感染基因型 3 HCV,1 名感染基因型 4 HCV),其中 35 名先前在接受基於干擾素的治療時治療失敗,16 名患有肝硬化。在 40 名受試者中,28 名 (70%) 實現 SVR12:22/33 名 (73%) 感染基因型 1 HCV 的受試者,6/6 名 (100%) 感染基因型 3 HCV 的受試者及 0/1 名 (0%) 感染基因型 4 HCV 的受試者。實現 SVR12 的所有受試者均實現 SVR24 和 SVR48。
不同治療方案和治療持續時間的結果概述-各研究比較
下列表格(表 21 至表 24)列出了 2 期和 3 期研究中的劑量相關數據,從而幫助臨床醫師判定個體患者的最佳方案。
表 21:不同治療方案和治療持續時間的結果-基因型 1 HCV 感染的各研究比較
 索磷布韋片
索磷布韋片n = 實現 SVR12 應答的受試者人數;N = 每組的受試者總數。
a. 對於先前接受過治療的 HCV 基因型 1 感染患者,尚無海外 3 期數據,但對於接受 Sovaldi、利巴韋林和聚乙二醇干擾素 α 聯合治療的中國受試者,已獲得相關數據(請參見【注意事項】和【臨床研究】)。
b. 治療這類患者時,應考慮到索磷布韋、聚乙二醇干擾素 α 和利巴韋林的治療持續時間可能需要超過 12 周,最長達 24 周;對於存在一項或多項曾與含干擾素的治療應答率較低相關的因素(先前對聚乙二醇干擾素 α 和利巴韋林治療無應答、晚期肝纖維化/肝硬化、基線病毒濃度高、黑色人種、IL28B 非 CC 基因型)的亞組,尤為如此。
c. 這些是探索性或 2 期研究。應謹慎解讀結果,因受試者數量較少,SVR 率可能會受到患者選擇的影響。
d. 兩項研究的匯總數據。
表 22:不同治療方案和治療持續時間的結果-基因型 2 HCV 感染的各研究比較
 索磷布韋片
索磷布韋片n = 實現 SVR12 應答的受試者人數;N = 每組的受試者總數。
a. 這些是探索性或 2 期研究。應謹慎解讀結果,因受試者數量較少,SVR 率可能會受到患者選擇的影響。
在 ELECTRON 研究中 (N = 11),與索磷布韋 + 利巴韋林聯用的聚乙二醇干擾素 α 的持續時間為 4–12 周。
b. 在這兩項研究中,所有患者都無肝硬化。
表 23:不同治療方案和治療持續時間的結果-基因型 3 HCV 感染的各研究比較
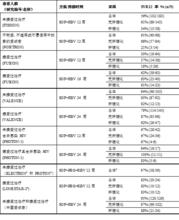 索磷布韋片
索磷布韋片n = 實現 SVR12 應答的受試者人數;N = 每組的受試者總數。
a. 這些是探索性或 2 期研究。應謹慎解讀結果,因受試者數量較少,SVR 率可能會受到患者選擇的影響。
在 ELECTRON 研究中 (N = 11),與索磷布韋 + 利巴韋林聯用的聚乙二醇干擾素 α 的持續時間為 4–12 周。
b. 在這兩項研究中,所有患者都無肝硬化。
表 24:不同治療方案和治療持續時間的結果-基因型 4、5 和 6 HCV 感染的各研究比較
 索磷布韋片
索磷布韋片兒童人群
在一項納入 50 名基因型 2 (n = 13) 或基因型 3 (n = 37) 慢性 HCV 感染患者的 2 期開放標籤臨床試驗中評估了索磷布韋在 12 至 < 18 歲 HCV 感染患者中的療效。該試驗中的 HCV 基因型 2 或 3 感染患者分別接受 12 或 24 周索磷布韋聯合利巴韋林治療。
在 50 名接受治療的患者中,中位年齡為 15 歲(範圍:12 至 17 歲);42% 的患者為女性;90% 為白人;4% 為黑人;2% 為亞裔;4% 為西班牙裔/拉丁裔;平均身體質量指數為 22 kg/m2(範圍:16 至 32 kg/m2);18% 接受過治療;66% 的基線 HCV RNA 水平大於或等於 800,000 IU/mL;74% 的患者攜帶非-CC IL28B 等位基因(CT 或 TT);無患者患有已知的肝硬化。多數患者 (69%) 通過垂直傳播感染。
基因型 2 患者的 SVR12 率為 100% (13/13),基因型 3 患者的 SVR12 率為 97% (36/37)。無患者出現治療期病毒學失敗或復發。1名基因型 3 HCV 感染患者實現 SVR4 但並未返回接受 SVR12 訪視。
藥理毒理
藥理作用:
作用機制
索磷布韋是 HCV NS5B RNA 依賴性 RNA 聚合酶(為病毒複製所必需)抑制劑。索磷布韋是一種核苷酸前體藥物,在細胞內代謝為具有藥理活性的尿苷類似物三磷酸鹽 (GS 461203),可被 NS5B 聚合酶嵌入 HCV RNA 中而終止複製。一項生化分析結果顯示,GS-461203 對基因型 1b、2a、3a 和 4a HCV 的重組 NS5B 的聚合酶活性具有抑制作用,50% 抑制濃度 (IC50) 為 0.7 ~ 2.6 μM 。GS-461203既不是人類 DNA 和 RNA聚合酶抑制劑,也不是線粒體 RNA 聚合酶抑制劑。
抗病毒活性
HCV 複製子分析結果顯示,索磷布韋對基因型 1a、1b、2a、3a 與 4a 中全長複製子的有效濃度 (EC50) 值分別為 0.04、0.11、0.05、0.05 和 0.04 μM,且索磷布韋對基因型 2b、5a 或 6a 中編碼 NS5B 的嵌合 1b 複製子的 EC50 值為 0.014 至 0.015 μM。對於基因型 1a、基因型 1b、基因型 2 和基因型 3a 臨床分離病毒株,索磷布韋對編碼 NS5B 序列的嵌合複製子的平均值 ± SD EC50 分別為 0.068 ± 0.024 μM (n = 67)、0.11 ± 0.029 μM (n = 29)、0.035 ± 0.018 μM (n = 15) 和 0.085 ± 0.034 μM (n = 106)。這些分析結果顯示,索磷布韋對較不常見基因型 4、5 和 6 的體外抗病毒活性與在基因型 1、2 和 3 中觀察到的抗病毒活性類似。40% 人血清對索磷布韋的抗 HCV 活性沒有影響。
耐藥性:
在細胞培養中
在包括 1b、2a、2b、3a、4a、5a 和 6a 等多種基因型的細胞培養物中,已選出對索磷布韋敏感性降低的 HCV 複製子。在檢查的所有複製子基因型中,對索磷布韋的敏感性降低均與原發性 NS5B 取代 S282T 有關。與相應的野生型相比,在 8 種基因型的複製子中,S282T 取代的定點誘變致使對索磷布韋的敏感性降低 2 至 18 倍,並使病毒複製能力降低 89% 至 99%。在生化分析中,與相應的野生型相比,來自表達 S282T 取代的基因型 1b、2a、3a 和 4a 的重組 NS5B 聚合酶顯示對 GS-461203 的敏感性降低。
在臨床試驗中
在對III期臨床試驗中接受索磷布韋治療的 991 名受試者進行的匯總分析中,226 名受試者因病毒學失敗或提前停用研究藥物且 HCV RNA > 1,000 IU/mL 而符合耐藥性分析資格。在這 226 名受試者中,獲得了 225 名的基線後 NS5B 序列,且獲得了其中 221 名受試者的深度測序數據(分析閾值為 1%)。在所有這些受試者中,通過深度測序或群體測序均未檢測到與索磷布韋相關的耐藥性取代 S282T。只在II期臨床試驗中接受索磷布韋單藥治療的一名受試者中檢測到 NS5B 的 S282T 取代。該受試者在基線時隱含 1% 的 HCV S282T,在治療後第 4 周時出現 S282T (> 99%),導致索磷布韋 EC50 發生 13.5 倍變化且病毒複製能力下降。在接下來的 8 周內 S282T 取代恢復為野生型,治療後 12 周通過深度測序無法再檢測到。
在III期臨床試驗中從多名基因型 3 HCV 感染受試者的治療後復發樣品中檢測到兩種 NS5B 取代,即 L159F 和 V321A。在出現這些取代的受試者分離病毒株中未檢測到對索磷布韋或利巴韋林的表型敏感性變化。此外,治療中,通過深度測序在一位出現部分治療應答的等待移植受試者中檢測到 S282R 和 L320F 取代。這些檢查結果的臨床意義尚不明確。
基線 HCV 多態性對治療結局的影響:
通過群體測序獲得了III期臨床試驗中 1,292 名受試者的基線 NS5B 序列,且在獲得基線序列的任何受試者中均未檢測到 S282T 取代。在一項評估基線多態性對治療結局影響的分析中,觀察到基線時任何 HCV NS5B 變異的存在與治療結局之間沒有具有統計學意義的關聯。
交叉耐藥性:
表達索磷布韋相關耐藥性取代 S282T 的 HCV 複製子對其他類的抗 HCV 藥物具有完全敏感性。索磷布韋對與其他核苷抑制劑耐藥性相關的 NS5B 取代 L159F 和 L320F 仍保有活性。對於與不同作用機制的其他直接作用抗病毒藥物(如 NS5B 非核苷抑制劑、NS3 蛋白酶抑制劑和 NS5A 抑制劑)耐藥性相關的取代,索磷布韋具有充分活性。
毒理研究
遺傳毒性:
索磷布韋Ames試驗、人外周血淋巴細胞染色體畸變試驗、小鼠微核試驗結果均為陰性。
生殖毒性:
索磷布韋在劑量為20、100、500mg/kg/日時,對大鼠胚胎-胎仔發育或生育力未見影響,500mg/kg/日劑量下主要循環代謝產物(GS-331007)的暴露量約為人臨床劑量下暴露量的8倍。在最高劑量下,索磷布韋對大鼠(500mg/kg/日)和兔(300mg/kg/日)未見致畸作用。主要循環代謝產物(GS-331007)在妊娠大鼠和妊娠兔體內的暴露量隨給藥時間的增加而增加,分別相當於人臨床劑量暴露的5~10倍和12~28倍。索磷布韋主要循環代謝產物(GS-331007)可通過乳汁分泌,對胎仔未見影響。
致癌性:
在小鼠和大鼠2年致癌性試驗中,雌雄小鼠給藥劑量分別達600mg/kg/日和200mg/kg/日,雌雄大鼠的給藥劑量達750mg/kg/日,未見致癌性。索磷布韋主要循環代謝產物(GS-331007)在小鼠體內的暴露量分別相當於人臨床劑量暴露的7倍(雄)和30倍(雌),在大鼠體內的暴露量分別相當於人日給藥劑量暴露量的13倍(雄)和17倍(雌)。
藥代動力學
索磷布韋是一種可被廣泛代謝的核苷酸藥物前體。活性代謝產物在肝細胞中形成,未在血漿中觀測到。主要 (> 90%) 代謝產物 GS 331007 是非活性成分。它經由連續和平行的代謝途徑形成活性代謝產物。
吸收
在健康成年受試者和患有慢性C型肝炎病毒感染的受試者中評估了索磷布韋和主要循環代謝產物 GS 331007 的藥代動力學特性。口服給藥後,無論劑量水平如何,索磷布韋均被迅速吸收,在給藥後約 0.5 2 小時觀測到藥物血漿濃度峰值。給藥後 2 至 4 小時之間觀測到了 GS 331007 血漿濃度峰值。基於基因型 1 至 6 HCV 感染受試者 (n = 986) 中的群體藥代動力學分析得出,索磷布韋和 GS 331007 的穩態 AUC0 24 分別為 1010 ng·h/mL 和 7200 ng·h/mL。在感染 HCV 的受試者中,索磷布韋和 GS 331007 的 AUC0 24 分別比健康受試者 (n = 284) 高 57% 和低 39%。
根據中國受試者 (N = 389) 中的群體 PK 分析,索磷布韋和 GS-331007 的穩態 AUC0-24 分別為 1730 ng·h/mL 和 6460 ng·h/mL。
食物影響
與空腹狀態相比,在進食標準高脂肪餐的狀態下給予單劑量索磷布韋使得索磷布韋的吸收速率降低。索磷布韋的吸收程度約增加 1.8 倍,對峰值濃度的影響很小。進食高脂肪餐的狀態下,GS 331007 的暴露量無變化。
分布
索磷布韋不是肝臟攝取性轉運體(有機陰離子轉運多肽 (OATP) 1B1 或 1B3 和有機陽離子轉運體 (OCT) 1)的底物。儘管 GS 331007 通過腎小管主動分泌,但它不是腎轉運體(包括有機陰離子轉運體 (OAT) 1 或 3、OCT2、MRP2、P gp、BCRP 或 MATE1)的底物。索磷布韋和 GS 331007 不是藥物轉運體 P gp、BCRP、MRP2、BSEP、OATP1B1、OATP1B3 和 OCT1 的抑制劑。GS 331007 不是 OAT1、OCT2 和 MATE1 的抑制劑。
索磷布韋與人血漿蛋白質的結合率大約為 85%(體外數據),藥物濃度在 1 μg/mL 至 20 μg/mL 時,此結合率不受藥物濃度影響。在人血漿中,GS 331007 的蛋白結合率極低。向健康受試者給予單劑量 400 mg 【14C】 索磷布韋後,14C 放射性的血液-血漿比約為 0.7。
生物轉化
索磷布韋在肝臟中被廣泛代謝,形成具有藥理學活性的核苷類似物三磷酸 GS 461203。代謝活化途徑包括經人組織蛋白酶 A (CatA) 或羧酸酯酶 1 (CES1) 催化的羧基酯部分的連續水解和經組氨酸三聯體核苷結合蛋白 1 (HINT1) 進行的磷醯胺酯裂解,之後通過嘧啶核苷酸生物合成途徑進行磷酸化。脫磷酸作用形成核苷酸代謝產物 GS 331007,此物質不能被有效地再磷酸化,且缺乏體外抗 HCV 活性。索磷布韋和 GS 331007 不是 UGT1A1 或 CYP3A4、CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19 和 CYP2D6 酶的底物或抑制劑。
經口給予單劑量 400 mg 【14C】 索磷布韋後,索磷布韋和 GS 331007 分別約占藥物相關物質系統暴露量(經分子量調整的索磷布韋及其代謝產物 AUC 之和)的 4% 和 > 90%。
消除
經口給予單劑量 400 mg 【14C】 索磷布韋後,劑量的平均總回收率大於 92%,其中尿、糞便與呼氣中分別約回收了 80%、14% 與 2.5%。尿中回收的索磷布韋劑量大部分是 GS 331007 (78%),另有 3.5% 以索磷布韋的形式回收。此項數據顯示 GS 331007 的主要消除途徑是腎清除,其中大部分可被主動分泌。索磷布韋和 GS 331007 的終末半衰期中位值分別為 0.4 和 27 小時。
線性/非線性
在空腹狀態健康受試者中評估了索磷布韋及其主要代謝產物 GS 331007 的劑量線性關係。當劑量範圍在 200 mg–400 mg 時,索磷布韋和 GS 331007 AUC 基本與劑量成比例。
特殊人群中的藥代動力學
性別和種族
對於索磷布韋和 GS 331007,未發現由性別和種族引起的臨床相關藥代動力學差異。
老年人
HCV 感染受試者的群體藥代動力學分析顯示,在分析的年齡範圍(19 至 75 歲)內,年齡對索磷布韋和 GS 331007 暴露量無臨床相關影響。
腎功能損害
在輕度(eGFR ≥ 50 且< 80 mL/min/1.73 m2)、中度(eGFR ≥ 30 且< 50 mL/min/1.73 m2)和重度 (eGFR 30 mL/min/1.73 m2) 腎功能損害以及患有需要血液透析的 ESRD 的 HCV 陰性受試者中,於 400 mg 劑量索磷布韋單次給藥後研究了索磷布韋的藥代動力學。與腎功能正常 (eGFR > 80 mL/min/1.73m2) 的受試者相比,在輕度、中度和重度腎功能損害受試者中,索磷布韋 AUC0 inf 分別高出 61%、107% 和 171%,GS 331007 AUC0 inf 則分別高出 55%、88% 和 451%。與腎功能正常的受試者相比,對於 ESRD 受試者,當在血液透析前 1 小時給予索磷布韋時索磷布韋 AUC0 inf 高出 28%,而血液透析後 1 小時給予索磷布韋則高出 60%。無法可靠地測定 ESRD 受試者的 GS 331007 AUC0 inf。但是數據表明在 ESRD 受試者進行血液透析前 1 小時或透析後 1 小時給予 Sovaldi,GS 331007 暴露量至少比腎功能正常受試者分別高出 10 倍或 20 倍。
血液透析可有效去除(提取率 53%)主要循環代謝產物 GS 331007。4 小時的血液透析會除去約 18% 的給藥劑量。對於輕度或中度腎功能損害患者,無需調整劑量。尚未評估 Sovaldi 在重度腎功能損害或 ESRD 患者中的安全性(請參見【注意事項】)。
肝功能損害
對患有中度和重度肝功能損害(CPT B 級和 C 級)的 HCV 感染受試者進行 7 天 400 mg 索磷布韋給藥後,研究了索磷布韋的藥代動力學。與肝功能正常的受試者相比,中度和重度肝功能損害受試者的索磷布韋 AUC0 24 分別高出 126% 和 143%,GS 331007 AUC0 24 則分別高出 18% 和 9%。感染 HCV 受試者的群體藥代動力學分析表明,肝硬化對索磷布韋和 GS 331007 暴露量無臨床相關影響。在輕度、中度和重度肝功能損害患者中,不建議對索磷布韋進行劑量調整(請參見【用法用量】)。
兒童人群
12 至 < 18 歲青少年中索磷布韋和 GS-331007 的暴露量與 2/3 期研究中成人接受索磷布韋 (400 mg) 給藥後的暴露量相似。尚未確定索磷布韋和 GS 331007 在 < 12 歲的兒童患者中的藥代動力學。
藥代動力學/藥效學關係
研究顯示,在快速病毒學應答方面,療效與索磷布韋和 GS-331007 暴露量相關。然而在 400 mg 的治療劑量下,沒有證據表明這些實體能成為療效 (SVR12) 的通用替代指標。
貯藏
30°C 以下保存。
包裝
高密度聚乙烯 (HDPE) 瓶,配有帶感應活性鋁箔內襯的聚丙烯連續螺紋防兒童開啟瓶蓋,內含 28 片薄膜衣片並有矽膠乾燥劑和聚酯棉塞。
提供以下包裝規格:每盒內含有 1 瓶,每瓶含 28 片薄膜衣片。
有效期
36 個月
執行標準
進口註冊標準:JX20160007
