
抗菌作用機制
 惡唑烷酮類藥物
惡唑烷酮類藥物Eustice等發現DuP-721能明顯地抑制[3H]標記賴氨酸的整合,但對[3H]標記胸腺嘧啶和尿嘧啶的整合影響很小,提示 DuP-721的作用機制為抑制細菌蛋白質合成。DuP-721與作用於蛋白質合成延長階段的抗菌素,如氯黴素和四環素不同,而是在更早階段阻斷蛋白質的合成。進一步研究發現,細菌經DuP-721預處理後,細菌提取物不能以正常的mRNA為模板,但若有人工合 成的多聚GU而缺乏3’上游核糖體結合序列的模板存在,卻能起始翻譯,也不影響fmet -tRNA與起始密碼GUG的結合以及肽鏈的延長。由此估計DuP-721可能抑制了核糖體 30S亞基對mRNA3’端上游核糖體結合序列的識別。 Shinabarger等在研究中證實了惡唑烷酮類物質不作用於翻譯的延長和終止階段,不影響met-tRNA和fmet-tRNA的合成。在以後的研究中,Lin等用放射標記的[14C]利奈唑烷(linezolid,U-100766)測定出惡唑烷酮類藥物與核糖體的結合特異地發生在50S 亞基上,這種結合可被氯黴素、林可黴素等競爭性抑制。已知氯黴素、林可黴素作用機制為抑制肽酸轉移酶並能與23SrRNA中某些位點的核苷酸發生作用,從而阻止肽鏈延長,並能影響翻譯的終止階段。儘管惡唑烷酮在核糖體上與氯黴素、林可黴素有共同的結合位點,但實驗中惡唑烷酮類並未表現出可以抑制肽醯轉移酶或影響翻譯終止,因此 Lin等推測惡唑烷酮類結合於核糖體50S亞基靠近氯黴素、林可黴素結合的位置,並且靠近與30S亞基接觸的界面。這種結合妨礙了 30S起始複合物與50S亞基結合形成70S起始複合物。
Kloss等通過對耐利奈唑烷菌株的研究發現,耐藥株存在核糖體50S亞基23SrRNA V 區域的變異。利奈唑烷與核糖體50S亞基的結合位點就包含了23SrRNA V區域的中心環,這也正是構成核糖體肽酸轉移酶中心不可缺少的部分。很明顯,rRNA對惡唑烷酮類發揮作用來說是非常重要的。他們推論該類藥物影響在前起始複合物形成過程中,fmet-tR- NA與50S亞基間的相互結合。
惡唑烷酮類對完整的大腸桿菌沒有作用,但當大腸桿菌細胞壁通透性增強時卻具有抗菌活性,這是由於在完整的革蘭氏陰性桿菌細胞壁上存在外排泵,可把細胞內藥物主動外排到胞外,從而降低了細胞內藥物濃度。
抗菌活性
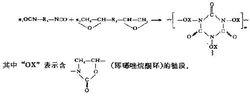 惡唑烷酮類分子式
惡唑烷酮類分子式體外抗菌活性
革蘭陽性菌:惡唑烷酮類除最早合成的S-6123抗菌活性較低外,其餘均顯示了較強的抗革蘭陽性球菌活性。早期合成物 DUP105、DuP721對金黃色葡萄球菌、β-溶血性鏈球菌、表皮葡萄球菌、腸球菌等的敏感性較強,其MIC90分別為8-16mg/L和2- 4mg/L。利奈唑烷和艾培唑烷(eperezolid, U-100592)對金黃色葡萄球菌的MIC90均≤ 4.0mg/L,對表皮葡萄球菌的MIC90分別≤2mg 和≤1mg/L。尤其值得注意的是,無論 MSSA、MSSE或MRSA、MRSE其抗菌活性均一致。利奈唑烷和艾培唑烷對鏈球菌屬,(包括化膿性鏈球菌)的MIC90分別為0.5- 4.0mg/L和0.5-2.0mg/L,且對紅黴素和/或青黴素耐藥的菌株其抗菌活性與敏感株相似。腸球菌對利奈唆烷和艾培唑烷也很敏感,對耐萬古黴素糞腸球菌的MIC90分別為 4.0mg/L和2.0mg/L,對耐萬古黴素屎腸球菌的MCI90/sub>均為2.0mg/L。利奈唑烷和艾培唑烷對軍團菌屬的MIC90均為4mg/L。新近開發的同類化合物PNU-171933、PNU- 172576對革蘭陽性菌表現出更強大的抗菌活性。它們對金黃色葡萄球菌、表皮葡萄球菌、肺炎鏈球菌和腸球菌的MIC均≤0.5mg/L。
革蘭陰性菌:惡唑烷酮類藥物對大多數革蘭陰性菌缺乏有效的抗菌活性,但PNU -171933、PNU-172576等分子中引入了氰化吡咯。氰化吡唑等基團,在對革蘭陽性菌具有強大抗菌活性的同時,對革蘭陰性菌也具有較強的抗菌活性。它們對流感嗜血桿菌的MIC均為4mg/L,對卡他莫拉菌的MIC分別為1mg/L和2mg/L。
厭氧菌:Jones研究了利奈唑烷和艾培唑烷對部分厭氧菌的抗菌活性。他們對脆弱擬桿菌、梭狀芽孢桿菌屬、消化鏈球菌屬等的MIC50為1-2mg/L,與克林黴素、甲硝唑作用相近。
體內抗菌活性
Ford等通過感染動物模型研究了利奈吐烷和艾塔唑烷的體內抗菌活性,利奈唑烷對MSSA和MRSA感染小鼠的 ED50分別為2.9-3.7mg/kg/天和2.8-15mg/ kg/天,艾培唑烷對MSSA和MRSA感染小鼠的ED50分別為5.6-6.9mg/kg/天和2.0-8.0 mg/lg/天。利奈唑烷和艾培唑烷對MRSE感染小鼠的ED504.7和1.9mg/kg/天。利奈唑烷對PHSP感染小鼠的ED50為2.5- 3.8mg/kg/天,其活性是頭孢克羅的5倍。艾培唑烷對PHSP感染小鼠的ED50為1.2- 11.7mg/kg/天。利奈唑烷對耐氨基糖甙類糞腸球菌和耐萬古黴素屎腸球菌感染小鼠的 ED50分別為10.0和24.0mg/kg/天,艾培唑烷對耐氨基糖甙類糞腸球菌和耐萬古黴素屎腸球菌感染小鼠的ED50分別為1.3和12.5mg/ kg/天。在小鼠多重耐藥肺炎鏈球菌性中耳炎中,利奈唑烷在炎住分泌物中的濃度達到血漿濃度的81%,在整個給藥期間,均高於對肺炎鏈球菌的MIC,且能夠滿意地消除中耳及鼻咽部的病原菌。在兔金黃色葡萄球菌性心內膜炎模型中,利奈唑烷按50或 75mg/kg口服給藥,血漿藥物峰谷藥物濃度分別為14.8與0.75倍MIC和24.7與5.8倍 MIC,均能明顯降低受感染免主動脈瓣疣狀贅生物計數值和瓣膜細菌培養陽性率。
利奈唑烷、艾培唆烷和PNU-100480對鼠結核分枝桿菌感染模型的抗菌活性研究表明,PNU-100480顯示出較強的抗菌活性,與異煙肼和/或利福平的抗菌活性相似,而利奈唆烷的抗菌活性較PNU-100480和異煙肼差,艾培唑烷僅有很弱的抗菌活性。
臨床研究
Chien等在一項非盲法、無對照的臨床試驗中,15例VRE感染患者,包括腹膜炎、菌血症、敗血症、肝膿腫、心內膜炎、直腸周圍膿腫等,並均伴有嚴重的基礎疾病如肝移植術後、HIV感染、糖尿病、白血病、腎功衰等,套用利奈唑烷600mgQ12H,療程5-42 天,平均20.5±3.5天。10例病員治療結束時病原體清除,7例獲臨床治癒,無一例死於VRE感染。
在一項有273例皮膚軟組織感染的患者參與的開放性臨床試驗中,靜脈或口服利奈唑烷,低劑量(250mg,每日3次;或375mg,每日2次)或高劑量(375mg,每日3次;或 625mg,每日2次)治療,靜脈給藥至少3 天,平均療程10天,隨訪15-28天。高、低劑量組的臨床治癒率分別達到87.7%和 83.3%,細菌清除率分別為90%和 82.5%。
在另一項門診病人社區獲得性肺炎治療試驗中,試驗組口服利奈唑烷600mg12H,對照組口服頭包泊肟200mgQ12H,治療結束後隨訪15-21天。試驗組201例可做臨床評價的病員中有180例治癒,治癒率為89.6%,其中肺炎鏈球菌、金黃色葡萄球菌、流感嗜血桿菌的清除率分別為92.6%、91.7%、 83.3%。治癒率和細菌清除率與對照組一致,提示利奈唑烷治療社區獲得性肺炎與頭孢泊肟同樣有效。
不良反應
Duvall總結了五項跨國多中心臨床試驗中利奈唑烷的不良反應,試驗組1498例患者每日靜脈或口服利奈唑烷600mgQ12H,對照組1464例患者所給藥物為克拉黴素、萬古黴素、頭孢曲松、頭孢泊肟、苯唑西林、雙氯西林等。試驗組不良事件發生率58.6%,對照組不良事件發生率52.4%,嚴重不良事件發生車試驗組和對照組分別為14.8%和 13.9%。最常見的與藥物相關的不良反應為腹瀉(試驗組4.0%,對照組2.7%)、噁心(試驗組3.3%,對照組1.8%)、頭疼(試驗組1.9%,對照組1.0%)。因與藥物相關的不良反應而終止試驗的試驗組有31人占 2.1%,對照組有25人占1.7%。患者對利奈唑烷顯示了良好的耐受性。
聯合用藥
惡唑烷酮類的抗菌譜主要為革蘭陽性菌。當存在革蘭陽性和陰性菌混合感染或尚未獲得細菌培養結果時,可同時配伍使用惡唑烷酮類及對革蘭陰性菌有效的藥物。Sisson 等研究了利奈唑烷與氨曲南在體內的相互作用,試驗結果未發現兩者聯合套用會出現有臨床意義的藥代動力學變化。在革蘭陽性菌和革蘭陰性菌混合感染的動物模型中,利奈唑烷、艾培唑烷與慶大黴素、氨曲南聯合用藥,可以有效發揮作用。
研究現狀
近年來,各類抗生素和抗菌藥的耐藥菌發展迅速,已嚴重威脅著感染性疾病患者的生命健康。例如:耐甲氧西林金黃色葡萄球菌(MRSA)、耐甲氧西林表皮葡萄球菌(MRSE)、耐青黴素肺炎鏈球菌(PRSP)、多重耐藥性結核桿菌,尤其是耐萬古黴素腸球菌(VRE)的出現,給臨床治療造成了困難。因此,探索新的抗耐藥性的革蘭氏陽性菌的藥物已成為國內外醫藥界的研究熱點。口惡唑烷酮類抗菌藥是繼磺胺類和氟喹諾酮類後的一類新型化學全合成抗菌藥,具有抑制多重耐藥革蘭氏陽性菌的作用。
1978年,美國杜邦公司報導了S-6123對細菌和真菌有活性後,又發現DUP105和DUP721對金黃色葡萄球菌等革蘭氏陽性菌有活性。1995年,美國Pharmacia&Upjohn公司以DUP721為先導化合物合成了依哌唑胺(Eperzolid)與利奈唑酮(linezolid),兩者對革蘭氏陽性菌具有和萬古黴素相似的抗菌活性。
利奈唑酮2000年4月在美國上市,主要用於治療由耐藥革蘭氏陽性菌引起的感染性疾病。該藥對骨骼、肺部、腦脊液等的滲透性和組織濃度的藥動學特徵良好,也可用於外科感染性疾病的治療。臨床前研究時,曾認為不太可能發生利奈唑酮耐藥。然而上市不到兩年,已有報導分離出耐利奈唑酮的葡萄球菌。而後臨床上又發現數株耐藥腸球菌。體外試驗提示,本類藥物耐藥突變株有可能通過改變核糖體的結合位點而產生耐藥性。因此人們對口惡唑烷酮類化合物進行了大量結構修飾或改造,期待獲得更具套用前景的抗菌藥物。
