
疾病概述
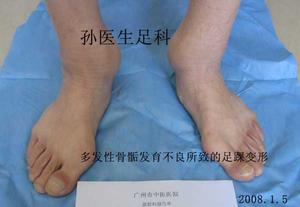 多發性骨骺發育不良
多發性骨骺發育不良多發性骨骺發育不良是少見的先天性骨發育障礙,多發性骨骺發育為常染色體顯性遺傳性骨病,僅侵犯骨骺軟骨,主要為軟骨發育過程中先期鈣化帶區的軟骨細胞未成熟,數量減少、排列不規整,致骨化障礙。多發性骨骺發育不良男女均可發病,約50%系家族性發病。
一般於4歲以後出現症狀,表現為關節疼痛,運動障礙和步態不穩。病變只侵犯及骨骺,以兒童時期最顯著,尤其以11-12歲症狀最明顯,青春期後隨年齡增長症狀可改善。
多發性骨骺發育不良嬰兒初生後並無明顯畸形,直至4—6歲走路不穩,橫距寬,個子矮小,方引起重視,青春期前可出現關節隱痛。多數病例只累及四肢,不累及脊柱與軀於,個別病例可出現脊柱側彎。四肢受累一般是對稱的,以髖、肩、膝、踝關節部位骨骺更為明顯。手指短粗,指甲短而鈍,重者握物能力明顯低下。身高最高不超過153cm。剝脫性骨軟骨炎,膝內、外翻等,但均無智力障礙。
當今醫學只有對多發性骨骺發育不良進行矯形,沒有特異治療,矯形手術不宜過早進行,骺末閉合前矯形,畸形復復發率甚高。
病因病理
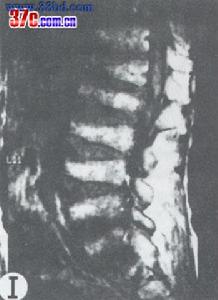 多發性骨骺發育不良--影像
多發性骨骺發育不良--影像多發性骨骺發育不良(MED)所具有特徵的性的骨骺停滯表現:平扁的、碎片的、均稱的骨骺的生成,矮小身材及早發骨性關節炎,為顯性基因遺傳,可找到突變的19號染色體,丹麥學者研究報導其患病率為9.0/100,000。
MED可發生在不同的位置,例如:椎體,鄰近的股骨通常也受累,易與幼年性變形性骨軟骨炎患病混淆,上肢較少累及,腕骨骨化中心形成延遲。MED是臨床及遺傳上的不良。6號染色體基因突變引起的MED已有報導。由軟骨低聚物基質蛋白(COMP),一種軟骨細胞中的a糖蛋白,可診斷MED。
病理學包含發育(骨)骺的骨化中心。軟骨內骨化是分裂的骨骺軟骨細胞不規則的。關節軟骨最初是正常的,但患者在病程中因為根本的骨性的支持鬆弛的,所以變得畸形。關節的畸形是永久的,在早期就顯示出成人的退化性變化及骨性關節炎。直到兒童晚期才能做出診斷。患兒長主訴關節僵直、疼痛、跛行像鴨步(態)。這樣的兒童身材矮小、肢體粗短。通常肘、膝部呈屈曲性攣縮,但智力是不受影響的。
臨床表現
 多發性骨骺發育不良
多發性骨骺發育不良多發骨骺發育不良分為嚴重型(Fairbank型)與輕型(Ribbing型)。但並不能包括所有的特徵,同一家族中,其表現也有所不同。輕的病例可因症狀不明顯不去就醫而不被發現,重的病例2—3歲就表現症狀,往往因行走延遲,行走不穩,身材特別矮小而就醫。根據Mena(1976)統計,在一個30名成員家族中,有6名受累,身高最高不超過153cm。
在受累家族中,非受累成員也可以出現骨關節畸形,如髖內翻,剝脫性骨軟骨炎,膝內、外翻等,但均無智力障礙。30—40歲以後,因退行性骨性關節炎改變症狀明顯加劇。
此外,還有的學者把其他類型骨骺發育不良也列入多發骨骺發育不良的範疇內。如Montry(1962)提出常染色體顯性遺傳雙髖骨骺發育不良,稱之為家族性股骨頭缺血壞死(familialPerthesdisease),其血緣發生率低於1%,雙親受累子女發生率為3%。Lowry與Wood(1975)報導兄弟二人多發骨骺不良,身材矮小,先天性眼震與小頭畸形,認為是性染色體或常染色體隱性遺傳。
x線表現
初生並無不良,股骨頭二次骨化中心出現延遲往往是最早的x線徵象,可以延遲到1—2歲才出現。股骨頭骺不規則,密度增加、不均勻、斑駁、破碎,很像Perthes病的表現。其他部位如股骨遠端,脛骨近端,脛骨遠端,肘、腕關節骨骺,均可有程度不等的變異,腕骨和跗骨間可發生融合。待發育成熟骨骺閉合後,關節面不規則,呈桑椹狀,股骨頸乾角減小,短頸、扁平髖,膝關節力線不良,內翻或外翻,踝關節踝穴變形,距骨滑車塌陷變扁,距骨頸變短,距骨頭扁平,距骨缺血壞死,跗骨扭曲,趾骨短縮,肘關節、腕關節間隙明顯變窄,尺撓骨發育不對稱,腕骨扭曲,掌骨變短。個別病例累及椎體,表現為椎體不規則,前方稍呈楔狀變形。
鑑別診斷
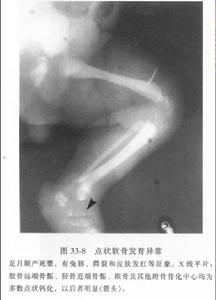 多發性骨骺發育不良
多發性骨骺發育不良主要與股骨頭缺血壞死(Perthes病)相鑑別,雙側受累,多關節受累是其主要鑒鑑別點。此外,還應與雙側短股骨、脛骨內翻、脊柱骨骺發育不良、假性脊柱骨骺發育不良等相鑑別。
臨床表現加之X線檢查除外Perthes病、先天性髖內翻Blount病和骨骺點狀發育不良可以診斷。股骨遠端骨骺高度與乾骺端寬度比值不良可見於多數患兒。這一指標對於早期診斷很有價值。
一般出生時無明顯不良2歲以後逐漸出現症狀走路較晚,步態不穩出現膝內、外翻關節疼痛,功能受限6-7歲時可出現脊柱側凸。四肢短身材矮小形如侏儒,但面部頭顱正常,智力發育不受影響。
辨證治療
 按摩揉推法
按摩揉推法2、按壓法:醫者雙手交叉按壓痛點1分鐘左右。
以上手法可循序進行。按壓後,術者雙手握住患者踝部,微用力做連續小幅度的上下牽抖10-20次左右而結束。手法治療不需每天都做,每周2次即可,連續治療2-3周。應該提醒患者的是,應去正規醫院的相關科室進行治療,以確保全全有效,不宜去非法行醫的個體按摩處求醫。手法治療對於接受過正規學習和訓練的操作者而言是安全有效的,但如果是沒有學習過解剖知識的個體按摩人員,難免粗野蠻幹,仍有一定的危險性。
3、拔罐法:用大、中號竹火罐閃火法自上往下,從患側臀部、下肢後外側拔閃罐至皮膚紅暈。再塗活絡油,拔循經走罐重複5-7遍。
4:溫和灸法:用藥艾條(蘇州市艾絨廠出品),循足太陽膀胱經、足少陽膽經自上而下,艾灸至能耐受為度,穴位周圍適當多灸。
5、針灸法(水針):患側臀部為主穴,秩邊、環跳、承扶、臀中、阿是穴每次取1-2個;風市、殷門交替取;陽陵泉、足三里、合陽、承筋每次取2個;藥用:VitB12ml、VitB22ml、10%GS15ml的混合液,每穴視肌肉豐厚情況注入1-3ml。每天1次,治療5次為1個療程,療程間休息2天,2個療程後統計療效。沒治療完2個療程又沒痊癒者,不作為本組病例,兩療程內痊癒者,作為痊癒統計。
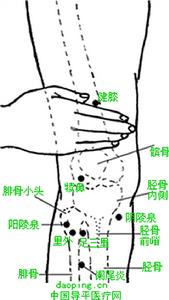 多發性骨骺發育不良
多發性骨骺發育不良(1)用掌或掌根沿梨狀肌走行及下肢後側肌施以推撫手法。
(2)單掌或掌根。拇指分別由匕至下揉梨狀肌5-7遍。
(3)用掌根以上至下揉大腿後側,至胭窩改為多指拿揉小腿後側三頭肌,反覆3-5遍。
(4)拇指撥揉坐骨神經路線3-5遍。
(5)肘尖撥壓梨狀肌2-3遍。
(6)雙拇指按梨狀肌走行撥理順壓3-5遍。
(7)雙手掌成掌根交替按壓下肢後側2-3遍。
(8)雙拇指交替按壓下肢後側坐骨神經路線3-5遍。
(9)掌指關節滾梨狀肌及下肢後側肌群3-5分鐘l0
(10)按壓環跳,承扶,殷門,委中,承山,昆倉穴每1-2分鐘。臀池(髂前上嵴與坐骨節結連線中點)以及局部壓痛點(阿是穴)。
(11)輕快地拿揉梨狀肌1-2分鐘,多指拿揉下肢後側2-3遍。
(12)輕叩或以拍打結束。
個案治療
一、治療方案
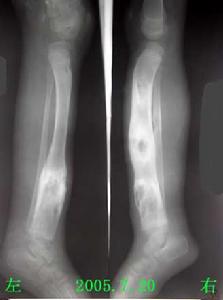
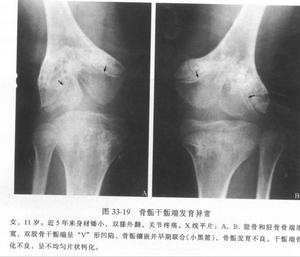 多發性骨骺發育不良--X片
多發性骨骺發育不良--X片多發性骨骺發育不良是少見的先天性骨發育障礙,本病為常染色體顯性遺傳性骨病,現將1例多發性骨骺發育不良報告如下。
患者,男,10歲,全身關節痛5年余,近1年來,全身關節疼痛更顯著,活動後易疲勞。其家族無其他人患此病。查體:身高106cm,體重26kg,智力尚正常,步態不穩。“X”型腿,雙手粗而短,指間關節腫脹,雙肘、雙膝、雙踝粗大,無紅熱,脊柱後凸,四肢與軀幹基本均稱。實驗室檢查:無不良發現。X線表現:兩側骨骺對稱性受累,骨骺發育小及變扁,可見呈分節或斑點狀,邊緣不規則,但無硬化帶。雙手掌指骨短,乾骺端粗大,雙舟骨不規則狀變形,各腕骨間骨間隙變窄。脊柱各椎體稍變扁,上下緣不規則且毛糙,頸椎下各
椎間隙稍變窄,腰椎第3、4、5椎體前後徑小,且胸腰段以胸11椎體至腰1椎體為中心後凸畸形。骨盆變窄,呈狹長型改變,雙股骨頭骨骺變扁平,並見有分節及斑點狀改變,股骨頸變短,髖臼變淺,雙膝關節間隙外寬內窄,雙側脛骨遠端骨骺外側部分發育不良,較細小,骨骺呈尖端指向外側之楔形,雙踝關節面傾斜。
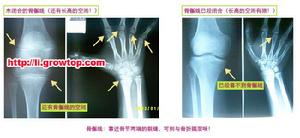 多發性骨骺發育不良
多發性骨骺發育不良多發性骨骺發育不良據資料於1921年首先被Barringten-Ward報導,1947年命名。本病僅侵犯骨骺軟骨,主要為軟骨發育過程中先期鈣化帶區的軟骨細胞未成熟,數量減少、排列不規整,致骨化障礙。本病男女均可發病,約50%系家族性發病。一般於4歲以後出現症狀,表現為關節疼痛,運動障礙和步態不穩。病變只侵犯及骨骺,以兒童時期最顯著,尤其以11-12歲症狀最明顯,青春期後隨年齡增長症狀可改善。身材較短小,四肢與軀幹基本勻稱,手足可粗短。表現為骨骺發育小而且變扁,邊緣不規則,可呈分節或斑點狀,但無硬化。管狀骨變短,關節面傾斜,脊柱後凸畸形,脛骨遠端骨骺有特徵性表現。隨年齡增長而乾骺端變小、扁平及關節畸形,較早地引起退行性骨關節病。本病須與軟骨發育不全、乾骺發育不良等骨關節發育障礙相鑑別。如能結合病史及其特徵性X線表現,是不難鑑別的。
