
基本原理
光譜的產生
在紫外光譜中,波長單位用nm(納米)表示。紫外光的波長範圍是10~380 nm,它分為兩個區段。波長在10~200 nm稱為遠紫外區,這種波長能夠被空氣中的氮、氧、二氧化碳和水所吸收,因此只能在真空中進行研究工作,故這個區域的吸收光譜稱真空紫外,由於技術要求很高,目前在有機化學中用途不大。波長在200~380 nm稱為近紫外區,一般的紫外光譜是指這一區域的吸收光譜。波長在400~750 nm範圍的稱為可見光譜。常用的分光光度計一般包括紫外及可見兩部分,波長在200~800 nm(或200~1000 nm)。
分子內部的運動有轉動、振動和電子運動,相應狀態的能量(狀態的本徵值)是量子化的,因此分子具有轉動能級、振動能級和電子能級。通常,分子處於低能量的基態,從外界吸收能量後,能引起分子能級的躍遷。電子能級的躍遷所需能量最大,大致在1~20 eV(電子伏特)之間。根據量子理論,相鄰能級間的能量差ΔE、電磁輻射的頻率ν、波長λ符合下面的關係式
ΔE=hν=h×c/λ
式中h是普朗克常量,為6.624×10⁻³⁴J·s=4.136×10⁻¹⁵ eV·s;c是光速,為2. 998×10¹⁰ cm/s。套用該公式可以計算出電子躍遷時吸收光的波長。
許多有機分子中的價電子躍遷,須吸收波長在200~1000 nm範圍內的光,恰好落在紫外-可見光區域。因此,紫外吸收光譜是由於分子中價電子的躍遷而產生的,也可以稱它為電子光譜。
躍遷類型
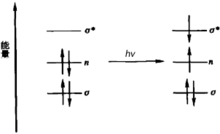
有機化合物分子中主要有三種電子:形成單鍵的σ電子、形成雙鍵的π電子、未成鍵的孤對電子,也稱n電子。基態時σ電子和π電子分別處在成鍵軌道和π成鍵軌道上,n電子處於非鍵軌道上。僅從能量的角度看,處於低能態的電子吸收合適的能量後,都可以躍遷到任一個較高能級的反鍵軌道上。躍遷的情況如下圖所示:
 各種電子躍遷的相對能量
各種電子躍遷的相對能量上圖中虛線下的數字是躍遷時吸收能量的大小順序,該順序也可以表示為
n→π*<π→π*><π→σ*><σ→π*><σ→σ*>
即n→π*的躍遷吸收能量最小。實際上,對於一個非共軛體系來講,所有這些可能的躍遷中,只有n→π*的躍遷的能量足夠小,相應的吸收光波長在200~800 nm範圍內,即落在近紫外-可見光區。其它的躍遷能量都太大,它們的吸收光波長均在200 nm以下,無法觀察到紫外光譜。但對於共軛體系的躍遷,它們的吸收光可以落在近紫外區。
根據上圖,可以認為:烷烴只有σ鍵,只能發生σ→σ*的躍遷。含有重鍵如C=C,C≡C,C=O,C=N等的化合物有σ鍵和π鍵,有可能發生σ→σ*,σ→π*,π→π*,π→σ*的躍遷。分子中含有氧、鹵素等原子時,因為它們含有n電子,還可能發生n→π*、n→σ*的躍遷。
一個允許的躍遷不僅要考慮能量的因素,還要符合動量守恆(躍遷過程中光量子的能量不轉變成振動的動能)、自鏇動量守恆(電子在躍遷過程中不發生自鏇翻轉),此外,還要受軌道對稱件的制約。即使是允許的躍遷,它們的躍遷機率也是不相等的。有機分子最常見的躍遷是σ→σ*,π→π*,n→σ*,n→π*的躍遷。
電子的躍遷可以分成三種類型:基態成鍵軌道上的電子躍遷到激發態的反鍵軌道稱為N→V躍遷,如σ→σ*,π→π*的躍遷。雜原子的孤對電子向反鍵軌道的躍遷稱為N→Q躍遷,如n→σ*,n→π*的躍遷。還有一種N→R躍遷,這是σ鍵電子逐步激發到各個高能級軌道上,最後變成分子離子的躍遷,發生在高真空紫外的遠端。
光譜圖
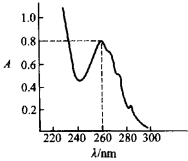 乙酸苯酯的紫外光譜圖
乙酸苯酯的紫外光譜圖右圖是乙酸苯酯的紫外光譜圖。
紫外光譜圖提供兩個重要的數據:吸收峰的位置和吸收光譜的吸收強度。從圖中可以看出,化合物對電磁輻射的吸收性質是通過一條吸收曲線來描述的。圖中以波長(單位nm)為橫坐標,它指示了吸收峰的位置在260 nm處。縱坐標指示了該吸收峰的吸收強度,吸光度為0.8。
吸收光譜的吸收強度是用Lambert(朗伯)—Beer(比爾)定律來描述的,這個定律可以用下面的公式來表示:
A=lg(I0/I)=kcl=lg(1/T)
式中A稱為吸光度(absorbance)。I0是入射光的強度,I是透過光的強度,T=I/I0為透射比(transmiπance),又稱為透光率或透過率,用百分數表示。l是光在溶液中經過的距離(一般為吸收池的長度)。c是吸收溶液的濃度。κ=A/(cl),稱為吸收係數(absorptivity)。若c以mol/L為單位,l以cm為單位,則κ稱為摩爾消光係數或摩爾吸收係數,單位為c㎡·mol(通常可省略)。
A,T,(1-T)(吸收率),κ,lgκ都能作為紫外光譜圖的縱坐標,但最常用的是κ,lgκ。上圖是以吸光度A為縱坐標的紫外光譜圖,下面四幅圖是以T,1-T,κ,lgκ為縱坐標的紫外光譜圖。由圖可知,透過率與吸收率正好相反,如吸收率為20%,透過率恰好為80%。
最大吸收時的波長(λmax)為紫外的吸收峰,在以吸光度、κ,lgκ、吸收率為縱坐標的譜圖中,λmax處於吸收曲線的最高峰頂,而在以透過率為縱坐標的譜圖中,λmax處於曲線的最低點。紫外吸收的強度通常都用最大吸收峰的κ值即κmax來衡量。在多數文獻報告中,並不繪製出紫外光譜圖,只是報導化合物最大吸收峰的波長及與之相應的摩爾消光係數。例如CH₃I的紫外吸收數據為λmax 258 nm(365),這表示吸收峰的波長為258 nm,相應的摩爾消光係數為365。
紫外光譜的測定大都是在溶液中進行的,繪製出的吸收帶大都是寬頻,這是 因為分子振動能級的能級差為0.05~1 eV,轉動能級的能差小於0.05 eV,都遠遠低於電子能級的能差,因此當電子能級改變時,振動能級和轉動能級也不可避免地會有變化,即電子光譜中不但包括電子躍遷產生的譜線,也有振動譜線和轉動譜線,解析度不高的儀器測出的譜圖,由於各種譜線密集在一起,往往只看到一個較寬的吸收帶。若紫外光譜在惰性溶劑的稀溶液或氣態中測定,則圖譜的吸收峰上因振動吸收而會表現出鋸齒狀精細結構。降低溫度可以減少振動和轉動對吸收帶的貢獻, 因此有時降溫可以使吸收帶呈現某種單峰式的電子躍遷。溶劑的極性對吸收帶的形狀也有影響,通常的規律是溶劑從非極性變到極性時,精細結構逐漸消失,圖譜趨向平滑。
電子躍遷
飽和有機化合物
飽和烴分子是只有C—C鍵與C—H鍵的分子,只能發生σ→σ*躍遷,由於σ電子不易激發,故躍遷需要的能量較大,即必須在波長較短的輻射照射下才能發生。如CH4的σ→σ*躍遷在125 nm,乙烷的σ→σ*躍遷在135 nm,其它飽和烴的吸收一般波長在150 nm左右,均在遠紫外區。
如果飽和烴中的氫被氧、氮、鹵素等原子或基團取代,這些原子中的n軌道的電子可以發生n→σ*躍遷。見下圖。
 n→σ*躍遷
n→σ*躍遷下表列舉了一些能進行躍遷的化合物。
| 化合物 | CH₃Cl | CH₃OH | CH₃OCH₃ | CH₃Br | CH₃NH₂ | CH₃I |
| λ max | 172(弱) | 183(150) | 185(2520) | 204(200) | 215(600) | 258(365) |
從上表可以看出,C—O(醇、醚),C-Cl等基團的n→σ*躍遷,吸收光的波長小於200 nm,在真空紫外,而C一Br,C一I,C-NH₂等基團的n→σ*躍遷,吸收光的波長大於200 nm,可以在近紫外區看到不強的吸收。這些化合物在吸收光譜上的差別,主要是由於原子的電負性不同,原子的電負性強,對電子控制牢,激發電子需要的能量大,吸收光的波長短;反之,原子的電負性較弱,對電子控制不牢,激發電子需要的能量較小,可以在近紫外區出現吸收。此外,分子的可極化性對其吸收光的波長也有一定的影響。可極化性大的,吸收光的波長也較長,n→σ*躍遷的κ值一般在幾百以下。
由於飽和烴、醇、醚等在近紫外區不產生吸收,一般用紫外-可見分光光度計無法測出,因此在紫外光譜中常用作溶劑。
不飽和脂肪族化合物
1.π→π*躍遷
C=C雙鍵可以發生π→π*躍遷,由於原子核對π電子的控制不如對σ電子牢,躍遷所需的能量較σ電子小。所以→π*躍遷κ值較大,在5000~100000左右,但是只有一個C=C雙鍵的躍遷出現在170~200 nm處,在真空紫外吸收,一般的分光光度計不能觀察到。例如乙烯的π→π*躍遷,λmax= 185 nm(κ=10000),在近紫外區不能檢出,同樣C≡C與C≡N等π→π*躍遷的吸收亦小於200 nm。
如果分子中存在兩個或兩個以上的雙鍵(包括三鍵)形成的共軛體系,π電子處在離域的分子軌道上,與定域軌道相比,占有電子的成鍵軌道的最高能級與未占有電子的反鍵軌道的最低能級的能差減小,使π→π*躍遷所需的能量減少,因此吸收向長波方向位移。消光係數也隨之增大,例如1,3-丁二烯分子中兩對π電子填滿π1與π2成鍵軌道,π3與T4反鍵軌道是空的,當電子吸收了所需的光能後便會發生從π2到π3的躍遷,見下圖。
由圖可知,在這種分子中,電子可以有多種躍遷,但是在有機分子中比較重要的是能量最低的躍遷,因為這種躍遷在近紫外區吸收,1,3-丁二烯的能量最低躍遷是π2→π3躍遷,其λmax=217 nm(κ= 21000),而其它躍遷能階相差較高,需要能量較大,在真空紫外吸收。隨著共軛體系逐漸增長,躍遷能階的能差逐漸變小,吸收愈向長波方向位移,由近紫外可以轉向可見光吸收(見下表)。
| 化合物 | 雙鍵 | λmax/nm( κ ) | 顏色 |
|---|---|---|---|
| 乙烯 | 1 | 185(10,000) | 無色 |
| 丁二烯 | 2 | 217(21,000) | 無色 |
| 1,3,5-己三烯 | 3 | 285(35,000) | 無色 |
| 癸五烯 | 5 | 335(118,000) | 淡黃 |
| 二氫-β-胡蘿蔔素 | 8 | 415(210,000) | 橙黃 |
| 番茄紅素 | 11 | 470(185,000) | 紅 |
因為共扼體系吸收帶的波長在近紫外,因此在紫外光譜的套用上,占有重要地位,對於判斷分子的結構,非常有用。
2.n→π*躍遷
有些基團存在雙鍵和孤電子對,如C=O,N=O,C=S,N=N等,這些基團除了可以進行π→π*躍遷,有較強的吸收外,還可進行n→π*躍遷,這種躍遷所需能量較少,可以在近紫外或可見光區有不太強的吸收,κ值一般在十到幾百。例如脂肪醛中C=O的π→π*躍遷吸收約210 nm,n→π*躍遷吸收約290 nm,見下左圖。
如果這些基團與C=C共軛,形成含有雜原子的共軛體系,與C=C—C=C共軛類似,可以形成新的成鍵軌道與反鍵軌道,使與π→π*與n→π*的躍遷能級的能差減小,吸收向長波方向位移,例如2-丁烯醛的π2→π3和n→π3躍遷與脂肪醛相應的躍遷比較,吸收均向長波位移,見下右圖。
 脂肪醛的π→π*和n→π*躍遷 脂肪醛的π→π*和n→π*躍遷 |  2-丁烯醛的π2→π3和n→π3躍遷 2-丁烯醛的π2→π3和n→π3躍遷 |
下表列舉了常見的n→π*躍遷化合物的吸收帶以及不同類型共軛分子的吸收帶。
| 化合物 | 基團 | π→π*λmax/nm( κ ) | n→π*λmax/nm( κ ) |
|---|---|---|---|
| 醛 | —CHO | ~210(強) | 285~295(10~30) |
| 酮 | 羰基 | ~195(1000) | 270~285() |
| 硫酮 | ~200(強) | ~400(弱) | |
| 硝基化合物 | —NO₂ | ~210(強) | ~270(10~20) |
| 亞硝酸酯 | —ONO | ~220(2000) | ~350(0~80) |
| 硝酸酯 | —ONO₂ | —— | ~270(10~20) |
| 2-丁烯醛 | CH₃=CHCH=O | ~217(16,000) | 321(20) |
| 聯乙醯 | O=CH-CH=O | —— | 435(18) |
| 2,4-己二烯醛 | CH₃=CHCH=CHCH=O | ~263(27,000) | —— |
從上表可以看出n→π*躍遷的值很小,一般是由十到幾百κ值小的原因,可以從羰基的軌道結構得到解釋(見右圖)。從圖中羰基的軌道圖中看到,n軌道的電子與π電子集中在不同的空間區域,因此,儘管n→π*躍遷需要的能量較低,由於在不同的空間,故n軌道的電子躍遷到π軌道的可能性是比較小的,產生躍遷的機率不大。由於κ值是由電子躍遷的機率決定的,所以n→π*躍遷的κ值很小,這種躍遷稱為禁忌躍遷,與n→π*躍遷比較κ值要小2~3個數量級。根據n→π*躍遷顯示弱的吸收帶,同時根據吸收位置,可以預示某些基團的存在,在結構測定中相當有用。
芳香族化合物
芳香族化合物都具有環狀的共軛體系,一般來講,它們都有三個吸收帶。芳香族化合物中最重要的是苯,苯的帶Ⅰλmax=184 nm(κ=47000),在真空紫外。帶Ⅱλmax=204 nm(κ=6900),帶Ⅲλmax=255 nm(κ=230)。下圖所示為苯的帶Ⅲ在255 nm處的吸收。因為電子躍遷時伴隨著振動能級的躍遷,因此將帶Ⅲ弱的吸收分裂成一系列的小峰,吸收最高處為一系列尖峰的中心,波長為255 nm,κ值為230,中間間隔為振動吸收,這種特徵可用於鑑別芳香化合物。
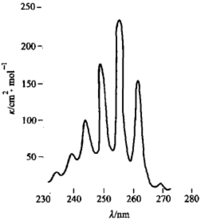 苯的紫外吸收光譜
苯的紫外吸收光譜苯衍生物的帶Ⅱ、帶Ⅲ亦均在近紫外吸收,下表是苯衍生物的吸收帶。
| 取代基 | 帶Ⅱλmax/nm( κ ) | 帶Ⅲλmax/nm( κ ) |
|---|---|---|
| H | 204(6,900) | 255(230) |
| -NH₃ | 203(7,500) | 254(160) |
| -CH₃ | 206(7,000) | 261(225) |
| —I | 207(7,000) | 257(700) |
| —Cl | 209(7,400) | 263(190) |
| —Br | 210(7,900) | 261(192) |
| —OH | 210(6,200) | 270(1,450) |
| —OCH₃ | 217(6,400) | 269(1,480) |
| -CO₂- | 224(8,700) | 268(560) |
| -COOH | 230(11,600) | 273(970) |
| -NH₂ | 230(8,600) | 287(1,430) |
| —O- | 235(9,400) | 287(2600) |
| -CHO | 244(15,000) | 280(1,500) |
| -CH=CH₂ | 244(12,000) | 282(450) |
| —NO₂ | 252(10,000) | 280(1,000) |
| 註:以上用水、甲醇或乙醇為溶劑。 | ||
有些基團的紫外吸收光譜與pH關係很大, 例如苯胺在酸性條件下由於氮上孤電子對與質子結合,它的吸收光譜與苯環類似;如酚在酸性與中性條件下的吸收光譜與鹼性時不一樣。
影響因素
生色基和助色基
凡是能在某一段光波內產生吸收的基團,就稱為這一段波長的生色基(chromophore)。紫外光譜的生色基是:碳碳共軛結構、含有雜原子的共軛結構、能進行n→π*躍遷的基團、能進行n→σ*躍遷並在近紫外區能吸收的原子或基團。常見的生色團列於下表。
| 生色團 | 化合物 | 溶劑 | λmax/nm | κ max |
|---|---|---|---|---|
| H₂C=CH₂ | 乙烯(或1-己烯) | 氣態(庚烷) | 171(180) | 15530(12500) |
| HC≡CH | 乙炔 | 氣態 | 173 | 6000 |
| H₂C=O | 乙醛 | 蒸汽 | 289,182 | 12.5,10000 |
| (CH₃)₂C=O | 丙酮 | 環己烷 | 190,279 | 1000,22 |
| -COOH | 乙酸 | 水 | 204 | 40 |
| -COCl | 乙醯氯 | 庚烷 | 240 | 34 |
| -COOC₂H₅ | 乙酸乙酯 | 水 | 204 | 60 |
| -CONH₂ | 乙醯胺 | 甲醇 | 295 | 160 |
| -NO₂ | 硝基甲烷 | 水 | 270 | 14 |
| (CH₃)₂C=N-OH | 丙酮肟 | 氣態 | 190,300 | 5000,— |
| CH₂=N⁺=N⁻ | 重氮甲烷 | 乙醚 | 417 | 7 |
| C₆H₆ | 苯 | 水 | 254,203.5 | 205,7400 |
| CH₃-C₆H₅ | 甲苯 | 水 | 261,206.5 | 225,7000 |
| H₂C=CH-CH=CH₂ | 1,3-丁二烯 | 正己烷 | 217 | 21000 |
| *孤立的C=C,C≡C的π→π*躍遷的吸收峰都在遠紫外區,但當分子中再引入一個與之共軛的不飽和鍵時,吸收就進人到紫外區,所以該表將C=C,C≡C也算作生色團。 | ||||
具有非鍵電子的原子連在雙鍵或共軛體系上,形成非鍵電子與π電子的共軛,即P-π共軛,使電子活動範圍增大,吸收向長波方向位移,並使顏色加深。這種效應,稱助色效應,這種基團稱為助色基(anxochmme),如一OH,一OR,一NH₂,一NR₂,一SR,鹵素等均是助色基。下表為乙烯體系、不飽和羰基體系及苯環體系被助色基取代後波長的增值。
| 體系 | NR2 | OR | SR | Cl | Br |
|---|---|---|---|---|---|
| X—C=C | 40 | 30 | 45 | 5 | — |
| X-C=C-C=O | 95 | 50 | 85 | 20 | 30 |
| X-C₆H₅ 帶Ⅱ | 51 | 20 | 55 | 10 | 10 |
| 帶Ⅲ | 45 | 17 | 23 | 2 | 6 |
| *表中X為助色基。 | |||||
紅與藍/紫移現象
由於取代基或溶劑的影響,使最大吸收峰向長波方向移動的現象稱為紅移(red shift)現象。由於取代基或溶劑的影響,使最大吸收峰向短波方向移動的現象稱為藍(紫)移(blue shift)現象。波長與電子躍遷前後所占據軌道的能量差成反比,因此,能引起能量差變化的因素如共軛效應、超共軛效應、空間位阻效應及溶劑效應等都可以產生紅移現象或紫移現象。
將烷基引入共軛體系時,烷基中的C一H鍵的電子可以與共軛體系的π電子重疊,產生超共軛效應,其結果使電子的活動範圍增大,吸收向長波方向位移。超共軛效應增長波長的作用不是很大,但對化合物結構的鑑定,還是有用的。下表列舉的數據表明了在共軛體系上的烷基對吸收波長的影響。
| 化合物 | λmax/nm |
|---|---|
| CH₂=CH-CH =CH₂ | 217 |
| CH₃-CH=CH-CH=CH₂ | 222 |
| CH₃-CH=CH-CH=CH-CH₃ | 227 |
| CH₂=C(CH₃)-C(CH₃)=CH₂ | 227 |
| CH₂=CH=C(CH₃)=O | 219 |
| CH₃-CH=CH-C(CH₃)=O | 224 |
| (CH₃)₂C=CH-C(CH₃)=O | 235 |
| C₆H₆ | 255 |
| CH₃-C₆H₅ | 261 |
由於溶劑與溶質分子間形成氫鍵、偶極極化等的影響,也可以使溶質吸收波長發生位移。如π→π*躍遷,激發態比基態的極性強,因此極性溶劑對激發態的作用比基態強,可使激發態的能量降低較多,以使基態與激發態之間的能級的能差減小,吸收向長波位移,即發生紅移現象。又如n→π*躍遷,在質子溶劑中,溶質氮或氧上的n軌道中的電子可以被質子溶劑質子化,質子化後的雜原子增加了吸電子的作用,吸引n軌道的電子更靠近核而能量降低,故基態分子的n軌道能量降低,n→π*躍遷時吸收的能量較前為大,這使吸收向短波位移,即發生紫移現象,見下圖。
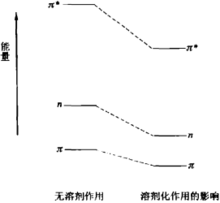 溶劑對溶質n→π*躍遷能量的影響
溶劑對溶質n→π*躍遷能量的影響由此可見,溶劑對基態、激發態與n態的作用是不同的,對吸收波長的影響亦不同,極性溶劑比非極性溶劑的影響大。因此在記錄吸收波長時,需要寫明所用的溶劑。紫外中常用的溶劑為水、甲醇,乙醇、己烷或環己烷、醚等。溶劑木身也有一定的吸收帶,雖然其κ值小,但濃度一般比待測物的濃度大好幾個數量級,因此,如果與溶質的吸收帶相同或相近,將會有干擾,選擇溶劑時,要予以注意。
增減色效應
使κ值增加的效應稱為增色效應(hyperchromic effect)。使κ值減弱的效應稱為減色效應(hypochromic cHect)。κ值與電子躍遷前後所占據軌道的能差及它們相互的位置有關,軌道間能差小,處於共平面時,電子的躍遷機率較大,κ值也就較大。在分子中,相鄰的生色基由於空間位阻效應而不能很好的 共平面,對化合物的吸收波長及κ值均有影響。例如二苯乙烯由於存在雙鍵,具有順反異構體,反式異構體的兩個苯環可以與烯的鍵共平面,形成一個大的共軛體系,它的紫外吸收峰在λmax=290 nm(k=27,000);而順式異構體兩個苯環在雙鍵的一邊,由於空間位阻不能很好地共平面,共軛作用不如反式的有效,它的紫外吸收λmax=280 nm(κ=14,000)。這種由於空間位阻使共軛體系不能很好共平面而引起的吸收波長與κ值的變化,在紫外吸收光譜中是一種普遍現象,在結構測定中十分有用。
套用範圍
醫藥方面
紫外光譜在破析一系列維生素、抗菌素及天然產物的化學結構曾起過重要作用,如維生素A1、維生素A2、維生素B12、維生素B1、青黴素、鏈黴素、土黴素、螢火蟲尾部的發光物質等。
例如利血平具有兩個共軛體系結構,水解得到利血平酸和3,4,5-三甲氧基苯甲酸。利血平酸經LiAlH4還原為利血平醇,其光譜與2,3-二甲基-6-甲氧基吲哚的紫外光譜相似。將合成的利血平醇與3,4,5-三甲氧基苯甲酸的紫外光譜疊加起來所得譜線與利血平的吸收曲線基本吻合,進一步由合成最後確定利血平的結構。
性能測試
光致變色現象是指在光的照射下顏色發生可逆變化的現象,可通過紫外光譜進行測試研究。如螺惡嗪類化合物A的環己烷溶液是沒有顏色,但在365nm連續的紫外光的照射下,溶液變成藍色,在可見區域產生吸收。隨照射時間的延長,吸收峰的強度逐漸變大,直至不再變化為止,將化合物的溶液放在暗處,其在可見光區域的吸收會逐漸下降。
光致變色材料作為一類新型功能材料,有著十分廣闊的套用前景。例如可以作為光信息存儲材料、光開關、光轉換器等,這些材料在機械、電子、紡織、國防等領域都大有作為。光致變色塗料、光致變色玻璃、光致變色墨水的研製和開發,具有現實性的套用意義。除了以上的套用,光致變色材料還可以作為自顯影感光 膠片、全息攝影材料、防護和裝飾材料、印刷版和印刷電路和偽裝材料等。
特別要指出的是,光致變色化合物作為可擦重寫光存儲材料的研究,是近些年來光致變色領域中研究的熱點之一。作為可擦寫光存儲材料的光致變色光存儲介質,應滿足在半導體雷射波長範圍具有吸收、非破壞性讀出、良好的熱穩定性、優良的抗疲勞性和較快的回響速度等條件。
