原因
發病原因
 男假兩性畸形
男假兩性畸形造成男假兩性畸形主要原因有:一是雄激素作用不全,其合成睪酮正常,但雄激素髮揮作用出現異常。二是睪丸激素合成缺陷,當間質細胞分化障礙或酶有遺傳性缺陷可導致46,XY的胎兒的中腎管男性生殖管道和外生殖器分化不完全,從而出現男假兩性畸形。三是Y染色體結構異常或基因突變,或者其他染色體異常導致性腺發育不全。
發病機制
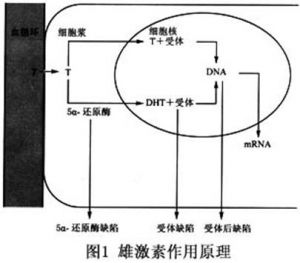
男假兩性畸形的發生機制與下列雄激素作用環節缺陷相關:①雄激素生成減少,如3β-HSD、17β-HSD和5α-還原酶缺陷;②雄激素受體(AR)基因突變,引起AR數量減少和功能障礙;③5α還原酶缺陷,不能將睪酮轉化為二氫睪酮(DHT);④靶細胞內雄激素代謝異常等,其中AR突變是引起完全型和不完全型睪丸女性化的主要原因。
人類雄激素受體(AR)基因位於Xq11~12,現已發現AR基因突變200餘種,其中基因單鹼基突變占90%以上。AR基因突變引起AR結合力降低和DNA結構異常,包括完全性基因缺失、編碼雄激素結合區或DNA結合區的外顯子缺失和點突變等。
1.完全型睪丸女性化 睪丸女性化患者的染色體性別是46,XY,常呈家族性發病,家系分析為X連鎖隱性遺傳病。在X染色體長臂上近著絲粒處-雄激素受體基因發生突變,靶細胞缺乏與雄激素結合的特異蛋白,雖存有生物活性的雄激素,但不與之結合而失去反應性。下丘腦也對雄激素不敏感而失去負反饋機制,導致垂體分泌大量促性腺激素刺激間質細胞增生。現已證實雄激素受體基因位於Xq11~12,長度大於90kb,有8個外顯子,外顯子1占據整個氨基端,具有激發轉錄的功能。睪丸女性化患者大多數是基因點突變或鹼基對的缺失,導致雄激素受體發生缺陷。致使男性外生殖器轉化受阻.
此類患者是由於靶組織缺乏雄激素受體,故對睪酮不敏感所致,是男假兩性畸形中最常見的類型,發生率在新生兒中約為1∶12萬。有家族發病史。睪丸在顯微鏡下像發育期前的隱睪,曲細精管變細,管內充滿支持細胞和未成熟的生精細胞,精子生成障礙,但間質細胞仍增生,這種睪丸易發生惡變。
2.不完全型睪丸女性化 又稱5α-還原酶缺乏症,遺傳學研究證實,患者染色體核型為46,XY。父母表型正常,發病隨血緣通婚而增加。家系分析大都可追溯到共同祖先患病。兩性都出現酶的異常缺陷,並且兩性都有表型正常的基因攜帶者,支持常染色體隱性遺傳。
已知在對雄激素敏感的靶組織中睪酮經5α-還原酶的作用,轉變為雙氫睪酮而起作用;而睪酮及雙氫睪酮在內生殖器向男性分化過程中是不可缺少的。若男性胎兒在發育早期缺乏5α-還原酶,則靶組織中雙氫睪酮缺乏,而引起外生殖器發育異常,常呈女性或男女難分。
根據5α-還原酶活性測定的結果,本病可分為酶缺乏和酶不穩定兩類。基因分析結果表明,酶活性缺乏的原因是基因的突變與缺失造成的,或者造成酶不能與睪酮結合,或者影響了酶的功能;或是酶編碼基因以外的突變影響基因的表達所致。
早期症狀
1.完全型睪丸女性化 患者的外生殖器完全和正常女性相同,陰蒂不肥大,但陰道比較淺,呈盲端。無子宮頸,腹腔內沒有子宮及輸卵管。原發性閉經。兩側睪丸的位置可在腹腔內、腹股溝部及大陰唇中,以腹股溝部最常見(78%)。兩側乳房發育肥大,但腺組織較少,乳頭髮育欠佳,體型也呈女性樣。本病的另一個特點是患者沒有腋毛及陰毛(無毛女人)。
2.不完全型睪丸女性化 患者生殖道、性腺、血漿性激素和促性腺激素、染色體組型、遺傳方式等均與完全性睪丸女性化症相同,但外陰部有不同程度的男性
 男假兩性畸形
男假兩性畸形化,伴陰毛及腋毛生長。這是因為靶組織雄激素受體的量或質低於正常水平而非完全缺乏,故有不同程度的男性化表現。
(1)Lubs綜合徵:患者部分中腎管發育,有性毛,呈男性體型,後聯合融合,外生殖器傾向於女性。
(2)Gilbert-Dreyfus綜合徵:患者呈男性體型,陰莖較小,伴有尿道下裂,部分中腎管發育,乳房肥大,男性化程度為中間型。
(3)Reifenstein綜合徵:男性外陰,陰莖短小,有不同程度的尿道下裂,陰囊分叉,發育期乳房肥大,有性毛,不育。
(4)Rosewater綜合徵:男性外陰部及生殖管,發育期乳房肥大,脂肪分布呈女性,有性毛,不育。
(5)假陰道會陰陰囊型尿道下裂:這類患者由於5-α還原酶缺陷,睪酮不能轉化為5-α雙氫睪酮,其結果是陰莖像陰蒂,尿道開口在會陰部,有一淺的陰道。中腎管像正常男性一樣分化,即有精囊、輸精管、附睪、射精管開口在生殖竇。睪丸在腹股溝或分叉的陰囊內。成年患者精液內可有成熟的精子及各類精細胞。在發育期患者出現男性第二性徵:肌肉發達,聲音低沉。乳房不發育,陰莖增大,能勃起和射精。激素測定示血睪酮值和正常男性相同,而5-α雙氫睪酮值降低。患者包皮、陰莖海綿體等組織中幾乎不能測到5-α雙氫睪酮,表明在這些組織中缺乏5-α還原酶,致使尿生殖竇及外陰部不能充分地向男性方向發育。根據患者家譜的分析,該患者的遺傳方式可能是常染色體隱性遺傳。
完全型睪丸女性化根據染色體核型46,XY、女性表型、子宮缺如、腹股溝疝、無陰毛、隱睪和男性血清睪酮測定可確定完全型睪丸女性化診斷。
不完全性睪丸女性化與完全型睪丸女性化基本相同,不同點在於出現部分男性化症狀和體徵。
檢查
1.完全型睪丸女性化生殖激素測定顯示,血清FSH和LH正常或輕度升高、睪酮濃度相當於男性成人水平、雌二醇濃度高於男性成人水平。睪丸活檢顯示,睪丸體積較小,白膜光滑,輸精管、附睪、曲細精管發育不全、管壁退化、有精原細胞,但無精子生成。間質細胞增生不良,稀疏地分布於曲細精管周圍。
2.不完全性睪丸女性化內分泌檢查 青春期患者睪酮水平與正常男性相似,主要為二氫睪酮水平低下,二者的比率增加,睪酮對二氫睪酮的轉變率降低。血漿促黃體素(LH)輕度上升。尿中17-羥皮質類固醇(17-HS)和17-酮皮質類固醇(17-KS)正常。
目前暫無相關資料。
與哪些疾病混淆
睪丸女性化綜合徵應予鑑別診斷的相關疾病包括:
1.副中腎管發育不全綜合徵(Mayer-Rokitansky-Kuster-Hauser綜合徵,MRH綜合徵) 即先天性無陰道畸形。MRH綜合徵染色體核型為46,XX、雙側性腺為卵巢、無陰道。但有陰毛和腋毛髮育、內生殖器為女性、血清雌激素濃度正常。
2.真兩性畸形 染色體核型為46,XX或46,XY或嵌合型、雙側性腺為睪丸/卵巢/卵睪、兩性內外生殖器發育、外陰畸形、表型為女性或男性或畸形。
3.Klinefelter綜合徵 即先天性睪丸發育不全,染色體核型為47,XXY可以鑑別。
兒童患者易與17β-羥脫氫酶缺乏症相混淆,後者屬睪酮生物合成缺陷,同樣具有假陰道、會陰、陰囊型尿道下裂症的特徵,但本病血漿睪酮正常,青春期有進行性男性發育,可排除睪酮生成不足。
注意事項
治療
1.完全型睪丸女性化 因為患者的外生殖器為完善的女性,並按女性撫養,故治療原則仍維持女性性別。治療上爭論點是什麼時候切除雙側睪丸,因為患者的睪丸易發生惡變,但這種睪丸在發育期能引起女性第二性徵的形成。目前認為,在20歲以前睪丸發生惡變較罕見,20歲以後惡變率逐漸增高,年過30歲以後,睪丸惡變發生率可高達25%左右。所以一般都主張在發育期後再將雙側睪丸切除,這樣既可使女性化更為完善,又可避免睪丸的惡性變。由於切除睪丸後患者常出現類似女性絕經期的症狀,如面色潮紅,乳房縮小,陰道上皮萎縮等,因此應長期給予雌激素來替代治療。對於陰道較淺或狹窄妨礙性交者,應在適當時延長或擴大。不宜告訴患者生殖腺為睪丸,只宜說不能生育,以免患者精神上遭受難以醫治的創傷。
2.不完全性睪丸女性化 不完全性睪丸女性化症的治療比完全性類型複雜,因其外陰部的變化比較大。如Reifenstein綜合徵及Rosewater綜合徵患者的外生殖器幾乎完全為男性,而且都當作男性撫育,治療以糾正尿道下裂,切除發育的乳房組織,補充男性激素即可。Lubs綜合徵因外生殖器傾向於女性,常常當作女性撫育,應根據性分化異常治療原則處理。對於假陰道會陰陰囊型尿道下裂,因患者在發育期出現明顯的男性第二性徵,診斷明確後,應當作男性給予相應的處置。
預後
目前暫無相關資料。
