
蛋白純化的一般原則
蛋白純化要利用不同蛋白間內在的相似性與差異,利用各種蛋白間的相似性來除去非蛋白物質的污染,而利用各蛋白質的差異將目的蛋白從其他蛋白中純化出來。每種蛋白間的大小、形狀、電荷、疏水性、溶解度和生物學活性都會有差異,利用這些差異可將蛋白從混合物如大腸桿菌裂解物中提取出來得到重組蛋白。
蛋白的純化大致分為粗分離階段和精細純化階段二個階段。一般蛋白純化採用的方法為樹脂法。粗分離階段主要將目的蛋白和其他細胞成分如DNA、RNA等分開,由於此時樣本體積大、成分雜,要求所用的樹脂高容量、高流速、顆粒大、粒徑分布寬.並可以迅速將蛋白與污染物分開,必要時可加入相應的保護劑(例如蛋白酶抑制劑),防止目的蛋白被降解。精細純化階段則需要更高的解析度,此階段是要把目的蛋白與那些分子量大小及理化性質接近的蛋白區分開來,要用更小的樹脂顆粒以提高解析度,常用離子交換柱和疏水柱,套用時要綜合考慮樹脂的選擇性和柱效兩個因素。選擇性指樹脂與目的蛋白結合的特異性,柱效則是指各蛋白成分逐個從樹脂上集中洗脫的能力,洗脫峰越窄,柱效越好。僅有好的選擇性,洗脫峰太寬,蛋白照樣不能有效分離。
程式
分離純化某一特定蛋白質的一般程式可以分為前處理、粗分級、細分級三步。
前處理
分離純化某種蛋白質,首先要把蛋白質從原來的組織或細胞中以溶解的狀態釋放出來並保持原來的天然狀態,不丟失生物活性。為此,動物材料應先剔除結締組織和脂肪組織,種子材料應先去殼甚至去種皮以免受單寧等物質的污染,油料種子最好先用低沸點的有機溶劑如乙醚等脫脂。然後根據不同的情況,選擇適當的方法,將組織和細胞破碎。動物組織和細胞可用電動搗碎機或勻漿機破碎或用超音波處理破碎。植物組織和細胞由於具有纖維素、半纖維素和果膠等物質組成的細胞壁,一般需要用石英砂或玻璃粉和適當的提取液一起研磨的方法或用纖維素酶處理也能達到目的。細菌細胞的破碎比較麻煩,因為整個細菌細胞壁的骨架實際上是一個借共價鍵連線而成的肽聚糖囊狀大分子,非常堅韌。破碎細菌細胞壁的常用方法有超音波破碎,與砂研磨、高壓擠壓或溶菌酶處理等。組織和細胞破碎後,選擇適當的緩衝液把所要的蛋白提取出來。細胞碎片等不溶物用離心或過濾的方法除去。
如果所要的蛋白主要集中在某一細胞組分,如細胞核、染色體、核糖體或可溶性細胞質等,則可利用差速離心的方法將它們分開,收集該細胞組分作為下步純化的材料。如果碰上所要蛋白是與細胞膜或膜質細胞器結合的,則必須利用超音波或去污劑使膜結構解聚,然後用適當介質提取。
粗分離
當蛋白質提取液(有時還雜有核酸、多糖之類)獲得後,選用一套適當的方法,將所要的蛋白與其他雜蛋白分離開來。一般這一步的分離用鹽析、等電點沉澱和有機溶劑分級分離等方法。這些方法的特點是簡便、處理量大,既能除去大量雜質,又能濃縮蛋白溶液。有些蛋白提取液體積較大,又不適於用沉澱或鹽析法濃縮,則可採用超過濾、凝膠過濾、冷凍真空乾燥或其他方法進行濃縮。
細分離
樣品經粗分級分離以後,一般體積較小,雜蛋白大部分已被除去。進一步純化,一般使用層析法包括凝膠過濾、離子交換層析、吸附層析以及親和層析等。必要時還可選擇電泳法,包括區帶電泳、等電點聚焦等作為最後的純化步驟。用於細分級分離的方法一般規模較小,但解析度很高。
結晶是蛋白質分離純化的最後步驟。儘管結晶過程並不能保證蛋白一定是均一的,但是只有某種蛋白在溶液中數量上占有優勢時才能形成結晶。結晶過程本身也伴隨著一定程度的純化,而重結晶又可除去少量夾雜的蛋白。由於結晶過程中從未發現過變性蛋白,因此蛋白的結晶不僅是純度的一個標誌,也是斷定製品處於天然狀態的有力指標。
離子層析法
蛋白純化離子交換層析法有劑和離子交換劑當被分離的蛋白質溶液流經離子交換層析柱時,帶有與離子交換劑相反電荷的蛋白質被吸附在離子交換劑上,隨後用改變pH或辦法將吸附的蛋白質洗脫下來。
有機溶劑提取
蛋白質純化有機溶劑提取的原理是:與水互溶的有機溶劑(如甲醇、乙醇)能使一些蛋白質在水中的溶解度顯著降低;而且在一定溫度、pH值和離子強度下,引起蛋白質沉澱的有機溶劑的濃度不同,因此,控制有機溶劑的濃度可以分離純化蛋白質。例如,在冰浴中磁力攪拌下,在4℃預冷的培養液中緩慢加入乙醇(-25℃),可以使冰核蛋白析出,從而純化冰核蛋白。由於在室溫下,有機溶劑不僅能引起蛋白質的沉澱,而且伴隨著變性。因此,通常要將有機溶劑冷卻,然後在不斷攪拌下加入有機溶劑防止局部濃度過高,蛋白質變性問題就可以很大程度上得到解決。對於一些和脂質結合比較牢固或分子中極性側鏈較多、不溶於水的蛋白質,可以用乙醇、丙酮和丁醇等有機溶劑提取,它們有一定的親水性和較強的親脂性,是理想的提取液。冷乙醇分離法提取免疫球蛋白最早由Cohn於1949年提出,用於製備丙種球蛋白。冷乙醇法也是WHO規程和中國生物製品規程推薦的方法,不僅解析度高、提純效果好、可同時分離多種成分,而且有抑菌、清除和滅病毒的作用。
藥物分離
一、根據蛋白質溶解度不同的分離
1、蛋白質的鹽析法:中性鹽對蛋白質的溶解度有顯著影響,一般在低鹽濃度下隨著鹽濃度升高,蛋白質的溶解度增加,此稱鹽溶;當鹽濃度繼續升高時,蛋白質的溶解度不同程度下降並先後析出,這種現象稱鹽析。
2、等電點沉澱法:蛋白質在靜電狀態時顆粒之間的靜電斥力最小,因而溶解度也最小,各種蛋白質的等電點有差別,可利用調節溶液的pH達到某一蛋白質的等電點使之沉澱,但此法很少單獨使用,可與鹽析法結合用。
二、根據蛋白質分子大小的差別的分離方法
1、透析與超濾:透析法是利用半透膜將分子大小不同的蛋白質分開。超濾法是利用高壓力或離心力,強使水和其他小的溶質分子通過半透膜,而蛋白質留在膜上,可選擇不同孔徑的瀘膜截留不同分子量的蛋白質。
2、凝膠過濾法: 也稱分子排阻層析或分子篩層析,這是根據分子大小分離蛋白質混合物最有效的方法之一。柱中最常用的填充材料是葡萄糖凝膠(Sephadex ged)和瓊脂糖凝膠(agarose gel)。
三、根據蛋白質帶電性質進行分離
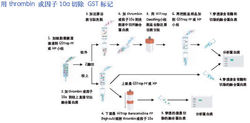 蛋白質純化流程
蛋白質純化流程1、電泳法:各種蛋白質在同一pH條件下,因分子量和電荷數量不同而在電場中的遷移率不同而得以分開。值得重視的是等電聚焦電泳,這是利用一種兩性電解質作為載體,電泳時兩性電解質形成一個由正極到負極逐漸增加的pH梯度,當帶一定電荷的蛋白質在其中泳動時,到達各自等電點的pH位置就停止,此法可用於分析和製備各種蛋白質。
2、離子交換層析法:離子交換劑有陽離子交換劑(如:羧甲基纖維素;CM-纖維素)和陰離子交換劑(二乙氨基乙基纖維素),當被分離的蛋白質溶液流經離子交換層析柱時,帶有與離子交換劑相反電荷的蛋白質被吸附在離子交換劑上,隨後用改變pH或離子強度辦法將吸附的蛋白質洗脫下來。
四、根據配體特異性的分離方法-親和色譜法
親和層析法(aflinity chromatography)是分離蛋白質的一種極為有效的方法,它經常只需經過一步處理即可使某種待提純的蛋白質從很複雜的蛋白質混合物中分離出來,而且純度很高。
這種方法是根據某些蛋白質與另一種稱為配體(Ligand)的分子能特異而非共價地結合。
其基本原理:蛋白質在組織或細胞中是以複雜的混合物形式存在,每種類型的細胞都含有上千種不同的蛋白質,因此蛋白質的分離(Separation),提純(Purification)和鑑定(Characterization)是生物化學中的重要的一部分,至今還沒的單獨或一套現成的方法能移把任何一種蛋白質從複雜的混合蛋白質中提取出來,因此往往採取幾種方法聯合使用。
主要方法
 蛋白質純化儀器
蛋白質純化儀器(1) 根據分子大小不同的分離方法:透析和超過濾(利用蛋白質分子不能通過半透膜的性質);密度梯度離心(蛋白質在介質中離心時質量和密度較大的顆粒沉降較快);凝膠過濾(一種柱層析)
(2) 利用溶解度差別分離:(等電點沉澱法)由於蛋白質分子在等電點時淨電荷為零,減少了分子間靜電斥力,因而容易聚集沉澱,此時溶解度最小);鹽溶與鹽析(利用一定濃度鹽溶液增大或減小蛋白質的溶解度)
(3) 根據電荷不同的分離方法,主要包括電泳和離子交換層析分離;
(4) 蛋白質的選擇吸附分離(利用顆粒吸附力的強弱不同達到分離目的)
(5) 根據配體特性的分離——親和層析(利用蛋白質分子與另一種稱為配體的分子能夠特異而非共價地結合這一生物性質)
(6)低溫有機溶劑沉澱法: 用與水可混溶的有機溶劑,甲醇,乙醇或丙酮,可使多數蛋白質溶解度降低並析出,此法分辨力比鹽析高,但蛋白質較易變性,應在低溫下進行。
注意事項
在進行任何一種蛋白質純化的時候,都要時刻注意維護它的穩定性,保護它的活性,有一些通用的注意事項需要牢記,它們包括:
1、操作儘可能置於冰上或者在冷庫內進行。
2、不要太稀,蛋白濃度維持在μg/mL~mg/mL。
3、合適的pH,除非是進行聚焦層析,所使用的緩衝溶液pH避免與pI相同,防止蛋白質的沉澱。
4、使用蛋白酶抑制劑,防止蛋白酶對目標蛋白的降解;在純化細胞中的蛋白質時,加入DNA酶,降解DNA,防止DNA對蛋白的污染。
5、避免樣品反覆凍融和劇烈攪動,以防蛋白質的變性。
6、緩衝溶液成分儘量模擬細胞內環境。
7、在緩衝溶液中加入0.1~1mmol/LDTT(二硫蘇糖醇)(或β-巰基乙醇),防止蛋白質的氧化。
8、加1~10mmol/LEDTA金屬螯合劑,防止重金屬對目標蛋白的破壞。
9、使用滅菌溶液,防止微生物生長。
內容提要
親和純化樣品的前處理
1. 菌液體積-起始目的蛋白量
2. 細菌裂解獲得可溶蛋白
· BugBuster Master Mix 蛋白抽提方法
· 機械破碎(如超聲等)Ni-NTA 樹脂純化
親和純化步驟
1. Ni-NTA樹脂的兼容性
2. 天然條件純化
· 柱層析
· FPLC
· 批次小量純化
3. 變性條件的純化
· 柱層析
· FPLC 純化
· 批次小量純化
4. 樹脂再生
5. 常見問題與改善建議
一、親和純化樣品的前處理
1. 菌液體積-起始目的蛋白量
純化條件的最佳化需考慮多個因素,包括His·Tag 融合蛋白表達水平和上樣量。若目的蛋白未能高效表達,需要采
用一個較高的濃縮係數(concentration factor)進行菌的裂解,即在較大培養體系中,按一定比例加入一定體積
的裂解/結合緩衝液。濃縮係數定義為菌液體積與裂解/結合緩衝液體積之比。不同目的蛋白表達水平推薦使用的
裂解/結合緩衝液體積、對應的濃縮係數大小,請參考表1。
例如,某蛋白表達水平約為0.1mg/ml,需要在變性條件下進行小批提純,則100ml 的培養物離心獲得的菌體,
按濃縮100 倍比例重懸於含變性劑的1ml 裂解/結合緩衝液中。
在非變性條件下進行純化時,要準確預計裂解液中可溶蛋白的含量比較困難,一般建議採用50-100 倍濃縮。
表1 確定所需菌液體積
His·Tag融合蛋白濃度表達水平培養體積 His·Tag融合
蛋白總量
濃縮係數
(在1ml溶液中裂解後)
變性條件
50mg/L 40% 3ml 150μg 3×
10mg/L 8% 10ml 100μg 10×
2mg/ L 1.6% 25ml 50μg 25×
0.5mg/ L 0.4% 50ml 25μg 50×
0.1mg/ L 0.8% 100ml 10g 100×
天然條件
>1mg/ L >1% 50ml >50g 50×
<1mg/ L <1% 100ml <100g 100×
與其它親和純化介質一樣,His·Bind 樹脂在接近其結合載量時使用,可以獲得最好蛋白分離效果。所以在蛋白純化前估計細菌
抽提物中目的蛋白的含量,有利於確定上樣量、選擇合適體積的親和樹脂或預裝柱。SDS-PAGE,Western blot,S·TagTM
Rapid Assay,FRETWorksTM S·Tag Assay 等方法都可用於確定抽提物中目的蛋白的含量。
2. 細菌裂解獲得可溶蛋白
· BugBuster Master Mix蛋白抽提方法
BugBuster Master Mix(貨號71456)是將BugBuster蛋白抽提試劑,Benzonase核酸酶和rLysozyme™溶菌酶溶
液按最最佳化配比混合好的一種非常方便使用的蛋白抽提試劑,有助於在最最佳化條件下獲得可溶活性蛋白。這種預
混形式的試劑減少了稀釋試劑、計算各種試劑使用量和分別添加的麻煩。100ml和500ml包裝分別足夠用於20g和
100g細胞沉澱的可溶蛋白抽提。
可溶蛋白製備
採用此操作抽提到的蛋白包括來自細胞周質和細胞質的可溶蛋白。如果欲僅收集周質部分蛋白,可以參考
Novagen的TB055“PET系統操作手冊”中提到的滲壓休克方法或其它合適的方法。滲透休克方法中得到的沉澱
也可以用於以下操作以獲得細胞質中的可溶蛋白。
1. 用經預先稱重的離心管10,000g離心10分鐘從液體培養體系收集細胞。如果是小規模培養(如1.5ml或更
少),可以用1.5ml離心管14,000–16,000g離心。儘量傾去液體,稱量細胞沉澱濕重。
2. 室溫下用吸打或溫和渦鏇使BugBuster Master Mix與細胞沉澱混勻,每克細胞糊需要5ml抽提試劑。這相當於
50ml培養液採用2.5ml抽提試劑。如果是小規模培養,則採用約1/5培養體積的抽提試劑重懸沉澱(例如,
1.5ml培養液採用300μl抽提試劑)。如果抽提試劑過量也沒有什麼副作用。
選做:加蛋白酶抑制劑。BugBuster Master Mix與蛋白酶抑制劑兼容。如果目的蛋白後續要用凝血酶(貨號
69671),Xa因子(貨號69036)或重組腸激酶(貨號69066)處理,就應該避免使用絲氨酸蛋白酶抑制劑。盡
管純化過程可能去除活性抑制劑,建議在酶切前最好做透析或凝膠過濾。
3. 室溫下將重懸的細胞液在搖板或低速攪拌器上孵育10-20分鐘。
注意:孵育後獲得的抽提物不是粘稠的。
4. 4℃下16,000g離心20分鐘以去除不溶的細胞碎片。如果需要,沉澱可以留作“包涵體純化”(見以下說明)
的材料。
5. 將上清轉入另一個新試管。這樣抽提得到的可溶蛋白溶液可以直接上樣於Novagen的純化樹脂(以及其它很
多類似純化系統)。蛋白溶液在冰上可以短時存放(2-3小時),也可在-20℃長時間存放直至下步分析。蛋
白抽提液應該根據目的蛋白的活性要求的溫度存放,有些蛋白經凍融會失活。
高純度包涵體的製備
以下操作可用於任何BugBuster®系列產品抽提的包涵體純化。
1. 如可溶蛋白抽提步驟1-4進行操作。
2. 將步驟“4”所得到的沉澱重懸於BugBuster(貨號70584),BugBuster的量與當初重懸細胞糊的體積相同。
吸打並渦鏇以獲得均勻的懸浮液。充分重懸沉澱能夠溶解、去除雜蛋白以獲得高純度的包涵體。
3. 加入rLysozyme™溶液至終濃度為1KU/ml。溫和渦鏇混勻,室溫孵育5分鐘。
注意:如果採用的是BugBuster Master Mix就不需要另加rLysozyme了(即可省去此步操作)。
4. 加入6倍體積的經1:10去離子水稀釋的BugBuster重懸,渦鏇1分鐘混勻。
5. 於4℃,5,000g離心15分鐘,以吸管移去上清,收集包涵體。
6. 將包涵體重懸於相當於原培養體系體積一半的經1:10稀釋的BugBuster中,渦鏇混勻,如步驟5離心。此步驟
重複兩次。再次重懸,於4℃,16,000g離心15分鐘並去除上清。
7. 重懸最終的沉澱(即純化的包涵體)於選定的緩衝液,最好是能與後續純化方法兼容的緩衝液。包涵體可以
重懸於Novagen的蛋白重摺疊試劑盒(貨號70123)中的1×溶解緩衝液(參考操作手冊TB234)或其它變性
劑。不溶的His•Tag®融合蛋白可以用含有變性劑的1×結合緩衝液重懸用於His•Bind®純化(IDA或NTA)。
機械破碎(如超聲等)
可溶蛋白的製備
稀釋8×儲液製成1×結合緩衝液,或按照緩衝液成分列表自己配製(參考Novagen 目錄或樹脂英文說明書)。
1. 10,000g 離心10 分鐘收集菌體。棄上清,儘量去除培養基。按每100mI 培養基所得菌體加入4ml (1:25 v/v)
緩衝液,重懸於冰浴預冷的1×結合緩衝液或1×Fractogel 結合緩衝液中。也可加入NP-40 或其他非離子型
去污劑至終濃度0.1%,以減少非特異性結合。若細菌菌體重懸困難,可使用勻漿器、攪拌器或超聲儀幫助打
散菌體。
2. 將重懸菌液置於合適大小的容器中,超聲破碎。超聲過程中保持菌液處於冰浴或鹽冰浴中。超聲條件依賴於
所使用的超聲儀功率、探頭種類、容器的大小形狀,須實驗者自己摸索。應避免連續超聲導致的大量產熱,
可分成短時間、多次超聲。通過一定的間隔時間避免溶液過熱。若DNA 未能被超聲剪下,裂解液會十分粘
稠,可能阻塞色譜柱,降低流速,影響後續的純化過程。大量的菌體可選用弗氏壓碎法(French Press)。
包涵體的製備
使用1×結合緩衝液從大腸桿菌中分離、洗滌包涵體,除去雜質蛋白,再用含6M 鹽酸胍或6M 尿素的1×結合緩
沖液溶解包涵體。
1. 10,000g 離心10 分鐘收集菌體。棄上清,儘量去除培養基。按每100mI 培養基所得菌體加入40mI 1×結合
緩衝液比例重懸菌體。此步驟不加變性劑。
2. 按前述方法進行超聲,重懸菌液,剪下降解核酸。
3. 5,000g 離心15 分鐘,包涵體和細胞碎片位於沉澱中。其他可溶蛋白部分位於上清中。
4. 移去上清,每100ml 培養物的沉澱重懸於20ml 1×結合緩衝液,重複第3 步。可能需要超聲以徹底重懸沉
淀。
注意: 本步操作可加入rLysozymeTM溶液(非必要)幫助處理包涵體。已證實溶菌酶可消化細胞壁,提高包涵體
純度。將rLysozyme 加入1×結合緩衝液至終濃度1KU/ml,輕柔混勻,孵育5-10 分鐘即可離心。
5. 移去上清,按每100ml 培養物加5mI 緩衝液比例,加入含6 M 鹽酸胍或6 M 尿素的1×結合緩衝液重懸沉
淀。
6. 冰浴孵育1 小時,徹底溶解包涵體。16,000g 離心30 分鐘去除不溶成分,His·Bind 純化之前用0.45um 濾膜
過濾上清。
二、親和純化步驟
Ni-NTA 樹脂純化
Ni-NTA His·Bind 樹脂不能耐受高濃度還原劑,如DTT、DTE,這些還原劑會還原Ni 離子,使其無法與His·Tag
融合蛋白結合。被還原的樹脂呈棕色。多數情況下,可以使用巰基乙醇,其濃度可以加至20mM。
EDTA、EGTA,或其它強螯合劑會與Ni2+結合,將其從樹脂上剝離下來。Ni2+被螯合後,樹脂呈白色。
對任何還原劑或螯合劑,請小心對待。若不能確定,請先在小量樹脂上進行試用。應儘量避免緩衝液中含有任何
高濃度供電子基團成分(如NH4+)、或裂解物中含諸如Arg、GIn、Gly、His 等胺基酸。
菌體應在無強螯合劑(如EDTA)、無強還原劑(如DTT)、無離子型去污劑(如SDS)的情況下裂解。儘管確實存在某
些例子,在這些試劑存在的情況下成功純化出目的蛋白,但還是建議大家避免使用這些試劑。
更多信息請參考2。
2. 天然條件純化
在決定使用天然條件(非變性條件)進行蛋白純化之前,首先需要確定蛋白是否可溶。即使目的蛋白大部分以不
溶形式表達,仍有可能有少量可溶蛋白可以用 Ni-NTA His·Bind 樹脂純化出來。
在沒有強變性劑如尿素的情況下,細胞裂解物中部分不穩定的蛋白可能容易被降解。最好始終保持蛋白溶液處於
低溫0-4℃,並快速操作。加入AEBSF 或蛋白酶抑制劑混合物系列III 貨號101500 和 539134),可能幫助蛋白
穩定(視具體情況而定),但是需要考慮到它們對重組蛋白的可能影響。
天然條件純化所需緩衝液(試劑盒70899)
1×Ni-NTA 結合緩衝液:(用於裂解和結合步驟)
50mM NaH2PO4,pH 8.0,300mM NaCl,10mM 咪唑
(加入1/10 體積的10×BugBuster 或溶菌酶能獲得更好裂解效果。參見前述“製備細菌裂解物”部分。)
1×Ni-NTA 漂洗緩衝液:50mM NaH2P04,pH 8.0,300mM NaCI,20mM 咪唑
1×Ni-NTA 洗脫緩衝液:50mM NaH2PO4,pH8.0,300mM NaCI,250mM 咪唑
柱
2. 天然條件純化
在決定使用天然條件(非變性條件)進行蛋白純化之前,首先需要確定蛋白是否可溶。即使目的蛋白大部分以不
溶形式表達,仍有可能有少量可溶蛋白可以用 Ni-NTA His·Bind 樹脂純化出來。
在沒有強變性劑如尿素的情況下,細胞裂解物中部分不穩定的蛋白可能容易被降解。最好始終保持蛋白溶液處於
低溫0-4℃,並快速操作。加入AEBSF 或蛋白酶抑制劑混合物系列III 貨號101500 和 539134),可能幫助蛋白
穩定(視具體情況而定),但是需要考慮到它們對重組蛋白的可能影響。
天然條件純化所需緩衝液(試劑盒70899)
1×Ni-NTA 結合緩衝液:(用於裂解和結合步驟)
50mM NaH2PO4,pH 8.0,300mM NaCl,10mM 咪唑
(加入1/10 體積的10×BugBuster 或溶菌酶能獲得更好裂解效果。參見前述“製備細菌裂解物”部分。)
1×Ni-NTA 漂洗緩衝液:50mM NaH2P04,pH 8.0,300mM NaCI,20mM 咪唑
1×Ni-NTA 洗脫緩衝液:50mM NaH2PO4,pH8.0,300mM NaCI,250mM 咪唑
柱
批次小量純化
實驗材料
·小離心管
·溶菌酶(貨號71110)
·Ni-NTA His·Bind 樹脂(貨號70666)
·1×Ni-NTA 結合緩衝液(緩衝液試劑盒貨號70899)
·1×Ni-NTA 漂洗緩衝液
·1×Ni-NTA 洗脫緩衝液
1、取1ml 菌液,加入一個小離心管中。
需要破碎多少菌體依賴於目的蛋白的表達水平。若蛋白高水平表達,1ml 菌液所含目的蛋白就足夠了(參考表
1)。若蛋白表達水平低,可能需要更大體積的菌液。若需要檢測誘導培養時間對蛋白表達水平的影響,每隔30
分鐘從培養體系中取出1ml 菌液,離心收集菌體,-20℃保存,直至所有樣品收集完全。
2、15,000g 離心1 分鐘,收集菌體,棄上清。
若需要更大體積的菌液,可向離心後去除上清的離心管中再加入菌液,再次離心,從而獲得更多菌體。
3、將細胞沉澱在-20C 冷凍15-20min。
4、將細胞完全解凍,用100μl 1×Ni-NTA 結合緩衝液重懸菌體。
若僅取1ml 菌液,濃縮係數為10,對於某些需要在天然條件下純化的蛋白來說可能不夠,請參考表 1 。
5、加入溶菌酶4.5-6KU 30 度孵育15min。
6、輕柔渦鏇,裂解細胞,避免起泡。
可選做:每100μl 重懸細胞加入2.5u Benzonase 核酸酶。可將Benzonase 稀釋10 倍(2.5u/ul)以方便取用
(稀釋緩衝液請垂詢)。
7、裂解物15,000g 離心10min。除去細胞不溶殘片,上清轉入乾淨小管中。
8、每管加入 20μl 50% Ni –NTA His·Bind 樹脂懸液(相當於 10μl 樹脂,可結合50-100μg His·Tag®融合蛋
白),4℃輕柔混勻,結合30 分鐘。
9、15,000g 離心10 s 沉澱樹脂,取 10μl 上清轉入另一乾淨小管中,以備電泳分析。棄去其它上清。
上清留樣保存於冰上。
3. 變性條件的純化
變性條件純化所需緩衝液
變性裂解/結合緩衝液
Buffer A:6M Gu-HCI;0.1M NaH2P04 ;0.01M Tris-CI,pH8.0
Buffer B:8M 尿素;0.1M NaH2P04;0.01M Tris-CI,pH8.0
變性漂洗緩衝液
Buffer C:8M 尿素;0.1M NaH2P04;0.01M Tris-CI pH6.3
變性洗脫緩衝液
Buffer D:8M 尿素;0.1M NaH2P04;0.01M Tris-CI pH5.9
Buffer E:8M 尿素;0.1M NaH2P04;0.01M Tris-CI pH4.5
注意:Buffer B、C、D、E應在使用前調至適宜pH值,以避免尿素解離。
柱層析
實驗材料
· 變性裂解/結合緩衝液裂解製備的細菌裂解物( 20-200ml 菌液體積)。
· Ni-NTA His·Bind 樹脂(貨號70666)
· 空色譜柱(貨號69673)
· Buffers A- E
1、將 1ml 50% Ni-NTA His·Bind 樹脂懸液加到 4ml 細胞裂解液中,輕柔混勻(如鏇轉混合器200rpm ) ,室
溫結合15-60min。
需要破碎多少菌體根據所使用的表達體系和His·Tag®融合蛋白的表達水平。Ni-NTA His·Bind 樹脂與不同的
His·Tag®融合蛋白的結合能力不同,通常為5-10 mg/ml 。如 Ni-NTA His·Bind 樹脂或Ni-NTA His·Bind
Superflow 對 His·Tag DHFR(~26 kDa)的結合量為 0.3μmol/ml (8.0 mg/ml )。請參考表 1。對於高表達水平的目的蛋白(每升培養基 50-100mg His·Tag 融合蛋白),可用5 倍濃縮的細菌裂解物(如
20ml 菌液離心獲得的菌體重懸於4ml Buffer B )。4ml 5 倍濃縮的裂解物含有約1-2mg 的 His·Tag 融合蛋白。
對於低表達水平的蛋白( 1-5 mg/1 培養基),可以50 倍濃縮,抽提 200ml 菌液,獲得 4ml 的細菌裂解物(4ml
細菌裂解物=0.2-1mg His·Tag 融合蛋白)。
2、將裂解液與Ni-NTA His·Bind 樹脂的混合物小心加入下端封閉的空色譜柱中。
3、除去柱下端封閉蓋子,收集流出液(穿過峰),保存用於SDS- PAGE 電泳分析。
4、以 4ml buffer C 漂洗雜蛋白2 次。
保存漂洗組分用於SDS- PAGE 電泳分析。
5、以 0.5ml buffer D 洗脫目的蛋白4 次,再以0.5ml buffer E 洗4 次,收集組分,用SDS-PAGE 分析。
蛋白單體通常用 buffer D 即可洗脫,而多聚體、聚合物和含兩個His·Tag 標籤的蛋白通常由buffer E 洗脫。
產品特點
1、處理過程為單純物理過程,無任何相變。設備操作溫度低,避免了傳統工藝的種種弊端;
2、系統採用先進的膜分離技術,工藝簡單,運行穩定可靠,處理效率高;
3、可以對生產廢水中的有用物質進行提純回用,實現經濟、環保雙贏;
4、設備投資少,運行費用低。
套用範圍
1、 化學物質的分離、提純、濃縮;
2、染料、染料中間體的濃縮及脫鹽
3、超細粉體生產過程中的產品回收;
4、生產廢水中有用物質的提純、回用;
5、海洋生物提取物的濃縮、提純
6、胺基酸、蛋白質的濃縮、提純。
