
範圍
本標準規定了原糧和成品糧中各項衛生指標的分析方法。
鎘、氟、曼陀羅籽、麥角、二溴乙烷、七氯、艾氏劑、狄氏劑等衛生指標的分析。
規範性引用檔案
下列檔案中的條款通過本標準的引用而成為本標準的條款。凡是注日期的引用檔案,其隨後所有的修改單(不包括勘誤的內容)或修訂版均不適用予本標準,然而,鼓勵根據本標準達成協定的各方研究是否可使用這些檔案的最新版本。凡是不注日期的引用檔案,其最新版本適用於本標準。
GB/T 5009.11 食品中總砷及無機砷的測定
GB/T 5009.15 食品中鍋的測定
GB/T 5009.17 食品中總汞及甲基汞的測定
GB/T 5009.18 食品中氟的測定
GB/T 5009.19 食品中六六六、滴滴涕殘留量的測定
GB/T 5009.20 食品中有機磷農藥殘留量的測定
GB/T 5009.22 食品中黃麴黴毒素的測定
感官檢查
具有正常糧食的色澤及氣味,不得有發霉變質現象。
理化檢驗
馬拉硫磷
氣相色譜法
按GB/T 5009.20操作。
銅絡合物比色法
原理
馬拉硫磷用有機溶劑提取,經氫氧化鈉水解後,生成二甲基二硫代磷酸酯,再與銅鹽生成黃色絡合物,與標準系列比較定量。
本方法取樣量為20 g時,檢出限為1.25 mg/kg。
試劑
四氯化碳。
無水乙醇。
硫酸鈉溶液(45 g/L):稱取4.5 9無水硫酸鈉,溶於水中,並稀釋至100 mL。
酸性硫酸鈉溶液:100 mL硫酸鈉溶液(45 g/L)中,加2.5mL鹽酸,混勻。
二硫化碳一四氯化碳混合液(1+200)。
鹽酸(1+1):取50 mL鹽酸,用水稀釋至100 mL。
氫氧化鈉溶液(240 g/L);取24 g氫氧化鈉,加水溶解至100 mL。
三氯化鐵溶液(50 g/L)。
硫酸銅溶液(35 g/L):稱取3.5g硫酸銅(CuS·5O)溶於100 ml。水中。
酚酞一乙醇指示液(10 g/L),稱取1 g酚酞,加乙醇(95%)溶解並稀釋至100 mL。
馬拉硫磷標準溶液:先將50 mL容量瓶準確稱量,然後滴入約50 mg馬拉硫磷,再準確稱量,加四氯化碳至刻度,混勻,並計算其濃度。
馬拉硫磷標準使用液:臨用前於馬拉硫磷標準溶液中加四氯化碳稀釋至每毫升相當於100.0馬拉硫磷。
儀器設備
分光光度計。
分析步驟
稱取約20. 00 g粉碎並全部通過20目篩的試樣,置於200 mL具塞錐形瓶中,加40 mL四氯化碳,密塞,振盪2h,然後過濾。
吸取20 mL濾液於125 mL分液漏斗中,加0.2 mL二硫化碳一四氯化碳混合液,加10 mL酸性硫酸鈉溶液,振搖1 mln靜置分層,將四氯化碳層轉入另一分液漏斗中,棄去水層。
吸取O、0.5、1.0、1.5、2.0、2.5 mL馬拉硫磷標準使用液(相當O、50、100、150、200、250 馬拉硫磷),分別置於125 mL分液漏斗中,加四氯化碳至20 mL,再各加0.2 mL二硫化碳一四氯化碳混合液。
於試樣溶液及馬拉硫磷標準溶液中各加5 mL無水乙醇,加0.4mL氫氧化鈉溶液(240 g/L),準確激烈振搖1 min,立即加入10 mL硫酸鈉溶液(45 g/L),混勻,加1滴酚酞指示液,用鹽酸(1+1)中和至酚酞將褪色,再用鹽酸(1+11)調pH為3~4(pH試紙試),再加0.5 mL三氯化鐵溶液(50 g/L)。振搖1 min,靜置分層(如乳化可離心分離),棄去四氯化碳層,用2 mE四氯化碳洗滌水層,振搖l min,分層後棄去四氯化碳層,如四氯化碳層帶黃色,再用四氯化碳洗滌水層。在水層中準確加入4.0 mL四氯化碳、0.5 mL硫酸銅溶液(35 g/L),準確振搖1 min。靜置分層後將四氯化碳層通過脫脂棉濾入2 cm比色杯中,以四氯化碳調節零點,在20 min內于波長415 nm處測吸光度,繪製標準曲線比較。
甲拌磷、殺螟硫磷、倍硫磷、敵敵畏、樂果、對硫磷
以氣相色譜法按GB/T 5009. 20操作。
磷化物
定性
原理
磷化物遇水和酸放出磷化氫,與硝酸銀生成黑色磷化銀,如有硫化物存在,同時放出硫化氫,與硝酸銀生成黑色硫化銀,干擾鋇4定,而硫化氫又能與乙酸鉛生成黑色硫化鉛,以此證明是否有硫化物干擾。
試劑
酒石酸。
硝酸銀溶液(100 g/L):貯存於棕色瓶中。
乙酸鉛溶液(loo g/L)。
乙酸鎘溶液(100 g/L)。
儀器
取2OO mL~250 mL錐形瓶,配一適宜雙孔軟木塞或橡皮塞,每孔內塞以內徑0.4 cm~0.5 em、長 5 cm的玻璃管,每管內懸掛一長7 cm、寬0.3 cm~0.5 cm的濾紙條,臨用時,一紙條用硝酸銀溶液濕潤,另一紙條用乙酸鉛溶液濕潤。
分析步驟
迅速稱取2O. 00 g試樣,置於錐形瓶中,加適量水至浸沒試樣,再加約0.5g酒石酸,立即塞好準備好的雙孔塞,使濾紙條末端距液面約5 cm,在暗處置40~50水浴內加熱30 min,觀察試紙顏色變化情況。如試紙均不變色,表明磷化物負反應或未超過規定;如硝酸銀試紙變色,乙酸鉛試紙不變色,表示
可能有磷化物存在,需再定量;如兩種紙均變色,可能有磷化物和硫化物同時存在或僅有硫化物存在,遇此情況,重取試樣,加水後再加5 mL乙酸鎘溶液(100 g/L),使形成黃色硫化鎘沉澱,立即密塞,放置10 min,再加酒石酸,操作同前,如硝酸銀試紙變黑,乙酸鉛試紙不變色,表示有磷化物存在,需再定量。
定量
原理
磷化物遇水和酸,放出磷化氫,蒸出後吸收於酸性高錳酸鉀溶液中被氧化成磷酸,與鉬酸銨作用生成磷鋁酸銨,遇氯化亞錫還原成藍色化合物鉬藍,與標準系列比較定量。
本方法取樣量為50 g時,檢出限0.020 mg/kg。
試劑
高錳酸鉀溶液(16.5 g/L):稱取16.5g高錳酸鉀,加水溶解後稀釋至1 000 mL,靜置三天或加熱煮沸3 min,冷卻,放置過夜,用玻璃棉或石棉過濾備用。
高錳酸鉀溶液(3.3 g/L):將高錳酸鉀溶液(16.5 g/L)用水稀釋5倍。
硫酸(1十17):取28 mL硫酸緩緩加入400 mL水中,冷卻後加水至500 mL。
硫酸(1+5):取83.3 mL硫酸緩緩加入400 mL水中,冷卻後加水至500 mL。
飽和亞硫酸鈉溶液:取28.5 g無水亞硫酸鈉,加約70 mL水,微熱溶勰後,放冷,稀釋至100 mL。
鉬酸錢溶液(50 g/L)。
氯化亞錫溶液;取0.lg氯化亞錫,溶於5 mL鹽酸中,臨用時現配。
磷化物標準溶液:準確稱取0.040 0 g經105乾燥過的無水磷酸二氫鉀,溶於水,移入100 mL容量瓶中,加水稀釋至刻度(可加一1滴三氯甲烷以增加保存時間),此溶液每毫升相當於0. 10 mg磷化氫。
磷化物標準使用液:吸取lO. OmL磷化物標準溶液,置於100 mL容量瓶中,加水至刻度。混勻-此溶液每毫升相當於10 磷化氫。
鹽酸(1+1)。
飽和硝酸汞溶液。
飽和硫酸肼溶液。
酸性高錳酸鉀溶液:高錳酸鉀溶液(16.5 g/L)和硫酸(2 mol/L)等量混合。
儀器
儀器如圖所示。
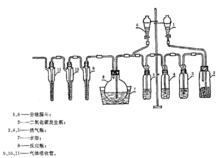 糧食衛生標準的分析方法
糧食衛生標準的分析方法分析步驟
試樣測定;於三個串聯的氣體吸收管中各加5 mL高錳酸鉀溶液(3.3 g/L)和1 mL硫酸(1+17)。二氧化碳發生瓶中裝大理石碎塊,從分液漏斗1中加適量的鹽酸(1+1),作為二氧化碳發生器。二氧化碳氣體順序經裝有飽和硝酸汞溶液、酸性高錳酸鉀溶液、飽和硫酸肼溶液的洗氣瓶洗滌後,進入反應瓶中(如用氮氣代替二氧化碳,可以只通過硫酸肼溶液安全瓶直接進入反應瓶)。預先通二氧化碳(或氮氣)5 min,打開反應瓶的塞子,迅速投入稱好的509試樣,立即塞好瓶塞,加大抽氣速度使分液漏斗6中的5 mL硫酸(1+17)和80 mL水加至反應瓶中,然後減慢抽氣和二氧化碳(或氮氣)氣流速度,將放置反應瓶的水浴加熱至沸30 min,並繼續通入二氧化碳(或氮氣)。反應完畢後,先除去氣體吸收管迸氣的一端,再除去抽氣管的一端,取下三個氣體吸收管,分別滴加飽和亞硫酸鈉溶液使高錳酸鉀溶液褪色,合併吸收管中的溶液至50 mL比色管中,氣體吸收管用少量水洗滌,洗液並人比色管中,加4.4mL硫酸(1+5),2.5 mL鋁酸銨溶液(50 g/L),混勻。
吸取0、0.1、0.2、0.3、0.4、0.5mL磷化物標準使用液(相當於0、1、2、3、4、5 磷化氫),分別放人50 mL比色管中,加30 mL水,5.4 mL硫酸(1-1-5),2.5 mL鉬酸銨溶液(50 g/L),混勻。予試樣及標準管中各加水至50 mL混勻,再各加0.1 mL氯化亞錫溶液,混勻。1S rain後。用3 cm比色杯,以零管調節零點,于波長680 nm處測吸光度,繪製標準曲線比較l或與標準系列目測比較。取與處理試樣量相同的試劑,按同一操作方法做試劑空白試驗。
結果計算
試樣中磷化物的含量按式(2)進行計算。
= (A1-A2)*1000/(m*1000)……………………………………(2)
式中:
——試樣中磷化物的含量(以磷化氫計),單位為毫克每千克(mg/kg);
——測定用試樣磷化物的質量,單位為微克();
——試劑空白中磷化物的質量,單位為微克();
——試樣質量,單位為克(g)。
計算結果保留兩位有效數字。
以空氣代替二氧化碳-空氣順序經裝有酸性高錳酸鉀溶液、鹼性焦性沒食子酸溶液(5 g焦性沒食子酸溶於15 mL水,48 g氫氧化鉀溶於32 mL水中,然後兩液混合)的洗氣瓶洗滌後進入反應瓶。以下操作同上。其裝置如圖所示。
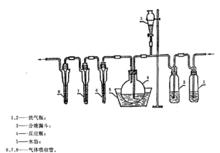 糧食衛生標準的分析方法
糧食衛生標準的分析方法氰化物
定性
原理
氰化物遇酸產生氫氰酸,氫氰酸與苦味酸鈉作用,生成紅色異氰紫酸鈉。
試劑
灑石酸。
碳酸鈉溶液(100 g/L)。
苦味酸試紙:取定性濾紙剪成長7 cm、寬0.3 cm~O.5 cm的紙條,浸入飽和苦昧酸-乙醇溶液中,數分鐘後取出,在空氣中陰乾,貯存備用。
儀器
取200 mL~300 mI,錐形瓶,配備一適宜的單孔軟木塞或橡皮塞,孔內塞以內徑0.4 cm~0.5 cm,長5 cm的玻璃管,管內懸一條苦味酸試紙,臨用時,試紙條以碳酸鈉溶液(100 g/L)濕潤。
分析步驟
迅速稱取5 g試樣,置於100 mL錐形瓶中,加20 mL水及0.5 g酒石酸,立即塞上懸有苦味酸並以
碳酸鈉濕潤的試紙條的木塞,置40~50水浴中,加熱30min,觀察試紙顏色變化。如試紙不變色,表示氰化物為負反應或未超過規定l如試紙變色,需再做定量試驗。
定母
原理
氰化物在酸性溶液中蒸出後被吸收於鹼性溶液中,在pH 7.0溶液中,用氯胺T將氰化物轉變為氯化氰,再與異煙酸一吡唑酮作用,生成藍色染料,與標準系列比較定量。
本方法取樣量為10 g時,檢出限為0. Ol5mg/kg。
試劑
甲基橙指示液(0.5 g/L)。
乙酸鋅溶液(100 g/L)。
酒石酸。
氫氧化鈉溶液(10 g/L)。
氫氧化鈉溶液(1 g/L)。
乙酸(1十24)。
酚酞一乙醇指示液(10 g/L)。
磷酸鹽緩衝溶液[(0.5 mol/L) pH7.O]:稱取34.0g元水磷酸二氫鉀和35.5 g無水磷酸氫二鈉,溶於水並稀釋至1 000 mL。
試銀靈(對二甲氨基亞苄基羅丹寧)溶液:稱取0.02 g試銀靈,溶於100 mL丙酮中。
異煙酸一吡唑酮溶液:稱取1.5g異煙酸溶於24 mL氫氧化鈉溶液(20 g/L)中,加水至100 mL,另稱取0.25g吡唑酮,溶於20 mLN-二甲基甲醯胺中,合併上述兩種溶液,混勻。
氯胺T溶液:稱取1 g氯胺T(有效氯含量應在11%以上),溶於100 mL水中,臨用時現配。
氰化鉀標準溶液:稱取0.25 g氰化鉀,溶於水中,並稀釋至1 000 mL,此溶液每毫升約相當於0.1 mg氰化物,其準確度可在使用前用下法標定。
取上述溶液10.O mL,置於錐形瓶中,加1 mL氫氧化鈉溶液(20 g/L),使pH為11以上,加0.1 mL試銀靈溶液,用硝酸銀標準溶液(0. 020 mol/L)滴定至橙紅色[1 mL硝酸銀標準溶液(0. 020 mol/L)相當於1.08 mg氫氰酸]。
氰化鉀標準使用液;根據氰化鉀標準溶液的濃度吸取適量,用氫氧化鈉溶液(1g/L)稀釋成每毫升相當於1 氫氰酸。
儀器
250mL玻璃水蒸氣蒸餾裝置。
分光光度計。
分析步驟
迅速稱取10.00 g試樣,放置於250 mL蒸餾瓶中,加適量水使試樣全部浸沒,加20 mL乙酸鋅溶液(100 g/L),加1 g~2 g酒石酸,迅速連線好全部裝置,冷凝管下端插入盛有5 mL氫氧化鈉溶液(10 g/L)的100 mL容量瓶的液面下,緩緩加熱,通水蒸氣進行蒸餾,收集餾液近100 mL,取下容量瓶,加水至刻度,混勻,取10 mL蒸餾液置於25 mL比色管中。
吸取O、0.3、0.6、0.9、1.2、1.5 mL氰化物標準溶液(相當於0、0.3、0.6、0.9、1.2、1.5 #g氫氰酸),分別置於25 mL比色管中,各加水至10 mL。於試樣溶液及標準溶液中各加1 mL氫氧化鈉溶液(10 g/L)和1滴酚酞指示液,用乙酸(1+24)調至紅色剛剮消失,加5 mL磷酸鹽緩衝溶液,加溫至37左右,再加入0.25 mL氯胺T溶液,加塞混合,放段5 min,然後加入5 mL異煙酸一吡唑酮溶液,加水至25 mL,混勻,於25℃"-,40℃放置40 min,用2 cm比色杯,以零管調節零點,予波長638 nm處測吸光度,繪製標準曲線比較。
結果計算
試樣中氰化物(以氫氰酸計)的含量按式(3)進行計算。
X= A*1000/(m*V1/v2*1000) ……………………………………(3)
式中:
——試樣中氰化物(以氫氰酸計)的含量,單位為毫克每千克(mg/kg);
——測定用試樣液氫氰酸的質量,單位為微克();
——試樣質量,單位為克(g);
——試樣蒸餾液總體積,單位為毫升(mL);
——測定用蒸餾液體積,單位為毫升(mL)。
計算結果保留兩位有效數字。
氯化苦
原理
氯化苦可被乙醇鈉分解形成亞硝酸鹽。在弱酸性溶液中與氨基苯磺酸進行重氮化,然後再與N-l-萘基乙二胺鹽酸偶合生成紫紅色,與標準系列比較定量。
本方法取樣量20 g時,檢出限為0.050 mg/kg。
試劑
乙醇鈉溶液:取金屬鈉,先用濾紙將表面煤油吸乾,並用小刀切去表面被氧化部分(切下表面部分,務必放回煤油中,切勿與水接觸),然後取5 g切成碎片,量取1 000 mL無水乙醇,置於大燒杯中,將切好的金屬鈉立即分次加入,待作用完畢,杯中不再有氣體發生時,移人棕色瓶中備用。
無水乙醇。
對氨基苯磺酸溶液(4 g/L):稱取0.4g對氨基苯磺酸溶於lOOmL鹽酸(1+1)中,貯存於棕色瓶中。
N-1-萘基乙二胺溶液(2 g/L)l稱取0.2 9 N-l-萘基乙二胺,溶於100m水中,貯存於棕色瓶中。
氯化苦標準儲備溶液:量取約20 mL無水乙醇,置於50 mL容量瓶中,準確稱量後,加入2滴氯化苦,再準確稱量,兩次的差即為氯化苦質量,加無水乙醇至刻度,混勻。
氯化苦標準使用液:吸取適量氯化苦標準儲備溶液,置於50 mL容量瓶中,加無水乙醇稀釋至刻度,此溶液每毫升應相當於0. 020 氯化苦,貯存於冰櫃中。
儀器
分光光度計。
分析步驟
稱取約20.00 g試樣,置於100mL具塞錐形瓶中,加20 mL乙醇鈉溶液,蓋好,放置暗處8 h~10 h或過夜,過濾,量取5.O mL濾液,置於10 mL比色管中。
吸取5.0mL氯化苦標準使用液a置於50 mL容量瓶中,加20 mL乙醇鈉溶液,放置暗處8 h~10 h或過夜,再加無水乙醇稀釋至刻度,然後吸取0、1.0、2.0、3.0、4.O、5.0 mL此液(相當於O、2、4、6、8、10 氯化苦),分別置於I0 mL比色管中,再各加元水乙醇至5 mL,加乙酸(36%)1 mL。
於試樣及標準管中各加1 mI。對氨基苯磺酸(4 g/L),混勻,靜置3 min~5 min後,各加入0.5 mLN-I-萘基乙二胺溶液(2 g/L)。加無水乙醇至刻度,混勻後放置20 min,用1 cm比色杯,以零管調節零點,于波長538 nm處測吸光度,繪製標準曲線比較。
4.5.5結果計算
試樣中氯化苦的含量按式(4)進行計算。
X= A*1000/(m*V1/v2*1000)……………………………………(4)
式中:
——試樣中氯化苦的含量,單位為毫克每千克(mg/kg);
——測定用試樣中氯化苦的質量,單位為微克();
——試樣中加入乙醇鈉總體積。單位為毫升(mL);
——測定用試樣濾液的體積,單位為毫升(mL);
——試樣質量,單位為克(g)。
計算結果保留兩位有效數字。
二硫化磷
原理
二硫化碳與二乙胺作用生成二乙胺磺酸,再與銅鹽反應生成黃色復鹽,與標準系列比較定量。
本方法取樣量為25 g時,檢出限為0. 20 mg/kg。
試劑
二乙胺-乙醇溶液(10 g/L)。
乙酸銅-乙醇溶液(0.5 g/L)。
硫酸銅溶液(50 g/L)。
二硫化碳標準儲備溶液:量取約20mL二乙胺一乙醇溶液(10 g/L),置於50 mL容量瓶中,準確稱量後,加入2滴二硫化碳,再準確稱量,兩次的差即為二硫化碳質量,加二乙胺一乙醇溶液(10 g/L)至刻度,混勻。
硫化碳標準使用液:吸取適量二硫化碳標準溶液,置於50 mL容量瓶中,加二乙胺一乙醇溶液(lO g/L)稀釋至刻度,此溶液每毫升相當於0.020 mg=硫化碳。
乙醇(95%)。
儀器
分光光度計。
分析步驟
稱取約26. 00 g試樣,置於600 mL圓底燒瓶中,加入水適量至浸沒試樣,於氣體吸收管內各加入110 mL二乙胺-乙醇溶液(10 g/L),洗氣瓶中加入50mL硫酸銅溶液(50 g/L),以除去空氣中的硫化物,三將圓底燒瓶置於70℃左右的水浴鍋中,按圖3連線好洗氣瓶、吸收管、抽氣裝置,緩緩抽氣2h後,取下氣體吸收管。停止加熱和抽氣。將氣體吸收管中吸收液倒入50 mL容量瓶中,用少量二乙胺一乙醇溶液(10 g/L)洗氣體吸收管
2次~3次,洗液併入容量瓶中,再用二乙胺一乙醇溶液(10 g/L)加至刻度,混勻。吸取5.0 mL置於125 mL比色管中。
吸取O、0.5、1.0、1.6、2.0、2.5mL一二硫化碳標準使用液(相當0、10、20、30、40、50二硫化碳),分別置於25 mL比色管中,再各加二乙胺·乙醇溶液(10 g/L)至10 mL,混勻。
於試樣及標準管中各加1 mL乙酸銅一乙醇溶液(0.5 g/L),再加乙醇至刻度,混勻。用1 cm比色杯,以零管調節零點,于波長400 nm處測吸光度,繪製標準曲線比較。
4.6.5結果計算
試樣中二硫化碳的含量按式(5)進行計算。
X= A*1000/(m*V1/v2*1000) ……………………………………(5)
式中:
——試樣中氯化苦的含量,單位為毫克每千克(mg/kg);
——測定用試樣中氯化苦的質量,單位為微克();
——試樣中加入乙醇鈉總體積。單位為毫升(mL);
——測定用試樣濾液的體積,單位為毫升(mL);
——試樣質量,單位為克(g)。
計算結果保留兩位有效數字。
砷
按GB/T 5009. 11操作。
汞
按GB/T5009. 17操作。
六六六、滴滴涕
按GB/T 5009.19操作。
黃麴黴薅素
按GB/T 5009.22操作。
鎘
按GB/T 5009.15操作。
氟
按GB/T 5009.18操作。
曼陀羅籽
鑑別
曼陀羅籽星三角形或腎形,扁平,表面有網點,呈棕色或棕褐色,有的邊緣有皺摺,淡棕色,寬2 mm~3 mm,有的較大,約5 mm~6 mm。
生物鹼比色定性
原理
試樣中所含阿托品等生物鹼經提取後與發煙硝酸及氫氧化鉀溶液有呈色反應。
試劑
氨水(1+1)。
乙醚。
鹽酸(1十5)。
三氯甲烷。
無水硫酸鈉。
發煙硝酸。
氫氧化鉀-乙醇溶液(100 g/L)。
分析步驟
將約30粒曼陀羅籽故人乳缽中,加氨水(1十1)浸濕,浸漬片刻,研磨成粘稠狀,加乙醚研磨三次,每次10 ml,,將乙醚合併於分液漏斗中,加10 mL鹽酸(1+5),振搖提取l min,分出鹽酸層至另一分液漏斗中,加氨水(1+1)調成鹼性,用10 mL三氯甲烷振搖提取1 min,再提一次,合併三氯甲烷層,通過無水硫酸鈉脫水後濃縮至0.5 mL,備用。
取0.2 mL試液於小蒸發皿中,揮乾溶劑,加4滴發煙硝酸使殘渣溶解,水浴上蒸於,殘留物變黃色,冷卻後加數滴氫氧化鉀一乙醇溶液(300 g/L),則變紫堇色,隨即變紅色。阿托品、莨菪鹼和東莨菪鹼均有此反應。
薄層色譜定性
原理
試樣中所含阿托品等生物鹼經提取後,用薄層分離,再以顯色劑顯色,與對照標準比較。
試劑
矽膠G薄層板:厚度0.3 mm~O. 5mm,105℃活化lh,放乾燥器中備用。
展開劑;甲醇-氨水(200十3)。
顯色劑:稱取0.85 g次硝酸鉍,加10 mL,冰乙酸,加40 mL水,溶解。取5 mL,加5 mL碘化鉀溶液(4 g碘化鉀溶於5 mL水中),再加20 mL冰乙酸,加水稀釋至100 mL。
阿托品標準溶液:稱取120.0 mg硫酸阿托品,溶於10 mL水中,加氨水(1十1)顯鹼性,用三氯甲烷提取二次,每次8 mL,三氯甲烷提取液經少許無水硫酸鈉脫水,濾人20 mL具塞比色管中,再用少許三氯甲烷洗濾器,洗液併入比色管中,加三氯甲烷至20 mL,此溶液每毫升相當於5.O mg阿托品。
東莨菪鹼標準溶液:稱取145.O mg氫溴酸東莨菪鹼,以下按阿托品標準溶液同樣處理,配成每毫升相當於5.0 mg東莨菪鹼。
分析步驟
在薄層板下端2 cm處,點10阿托品及東茛菪鹼標準溶液,30 ~lOO試樣提取濃縮液,各點間距1.5 cm,置於預先用展開劑飽和的展開槽中,待溶劑前沿上展至I0 cm~15 cm,取出,揮乾展開劑,噴顯色劑呈現橙紅色斑點為陽性反應。
定置
稱取1 000 g糧食,從中檢出曼陀羅籽,不得超過5粒。
麥角
鑑別
形態
麥角呈三條或四條鈍的圓柱形,微彎:兩端稍窄細,長0.3 cm~O.4 cm。粗1 mm~7 mm,外面呈黑色或紫棕色,有縱溝與橫裂紋,質脆,易折斷,斷面鈍三角形,邊緣暗紫色,中心呈灰白或紫白色。
組織切片
將麥角泡入水中,浸泡24 h,使膨腑夾在土豆或蘿蔔中間並固定,用小手術刀切成儘可能薄的小片,用次甲基藍溶液(1 g/L)顯色,在顯微鏡下觀察,其組織緊密。
麥角紅素和麥角生物鹼定性
試劑
酒石酸溶液(20 g/L)。
無水乙醚。
飽和碳酸氫鈉溶液。
氨水(1十1)。
三氯甲烷。
對二甲氨基苯甲醛溶液。稱取0.125 g對二甲氨基舉甲醛,加100 mL稀硫酸(65 mL硫酸緩緩倒入35 mL水中,混勻,冷卻)溶解,然後加0.1mL三氯化鐵溶液(50 g/L),混勻。
硫酸。
無水乙醇:紫外光燈波長365 nm下觀察無螢光。
乙酸乙酯。
過氧化氫溶液(3%)。
分析步驟
取20粒可疑麥角予研缽中,研碎,加酒石酸溶液(20 g/L)研成粘稠狀,加10 mL乙醚,仔細研磨,分取乙醚,反覆進行三次,合併乙醚層於試管中,保留殘渣於研缽內。在試管內加0.5 mL飽和碳酸氫鈉溶液,振搖,碳酸氫鈉溶液層變紅色,即表示檢出麥角紅。
取保留的殘渣,加氨水(1+1)研磨呈鹼性,用三氯甲烷提取三次,每次10 mL,合併三氯甲烷層,分成兩部分,一部分小心加2 mL對二甲氨基苯甲醛溶液,在兩液層接觸面呈藍紫色環,數分鐘後,三氯甲烷層均顯藍色,即表示檢出麥角生物鹼。另一部分三氯甲烷提取液置於試管中,在熱水浴上使三氯甲烷盡,殘留物加無水乙醇溶解,在波長365 nm紫外光燈下觀察有強烈藍色螢光,即表示檢出麥角生物鹼。
定量
稱l 000 g糧食,檢出麥角後稱量,不得超過0.lO g。
毒麥
根據形態鑑別,毒麥籽實被內外稃緊包。緊貼於內稃內,其芒聯接於外稃部,芒長7 mm~15 mm,籽實長橢圓型,堅硬無光澤,呈灰褐色,長4 mm~6 mm,腹溝較寬,籽實1 000粒,質量10g~13 g,
定量;稱取1 000 g糧食,揀出的毒麥不得超過1 g。
二溴乙烷
蒸餾法(SGS法)
試劑
己烷:重蒸,使用前以氣相色譜法檢驗,應無干擾蜂。
去泡劑(baker andifoam B)。
無水硫酸鈉。
二溴乙烷(EDB)標準使用液:用己烷配製成含1,0、2.5、5.0、10.0 ng/mL EDB的標準使用液。
儀器
氣相色譜儀:附有電子捕獲檢測器。
分析步驟
試樣處理
快速稱取50.0 g試樣(貯存子5℃以下)於1 000 mL圓底燒瓶中,加300 mL水、10.0 mL己烷,連線燒瓶、接收管、冷凝管,放在加熱器上緩緩加熱,用去泡劑避免過多的發泡[幾滴矽油或液體石蠟或吐溫(Tween) 80],加熱到已烷全部蒸出,水分開始在已烷層下集聚,移去加熱器,待其冷卻後,記下回收的己烷毫升數,並轉入具塞試管內,加2 9~3 9無水硫酸鈉脫水,供氣相色譜測定。
氣榻色譜參考條件
色譜柱;2 m×3 mm(內徑)不鏽鋼柱,內裝塗有10~% Squalene的80目~100目Chromosorb W HP。
溫度:柱溫:75℃;檢測器,275℃或300℃;進樣口:200℃;
載氣:氮氣50 mL/min。
測定
準確吸取2 #L樣液及不同濃度EDB標準使用液,注入氣相色譜儀中,每個試樣重複三次,取平均峰高,以標準濃度值對相應的色譜峰高值作EDB標準曲線,再以樣液峰高與標準曲線比較定量。
4.16.1.3.4結果計算
試樣中二溴乙烷的含量按式(6)進行計算。
X= A*1000/(m*V1/v2*1000) ……………………………………(6)
式中:
——試樣中二溴乙烷的含量,單位為微克每千克(mg/kg);
——進樣樣液相當二溴乙烷的質量,單位為納克();
——試樣進樣體積,單位為微升(mL);
——收集的己烷蒸餾液的體積,單位為毫升(mL);
——試樣質量,單位為克(g)。
計算結果保留兩位有效數字。
浸漬法
試劑
丙酮:重蒸,使用前以氣相色譜法檢驗,應無干擾峰。
無水氯化鈣。
氯化鈉。
丙酮-水(5+1)。
二溴乙烷標準使用液:用丙酮配製並稀釋至每毫升含0.2 二溴乙烷。
儀器
氣相色譜儀,附有電子捕獲檢測器。
分析步驟
試樣處理
快速稱取50.0 g試樣(貯存於5℃以下),置於250mL具塞錐形瓶中,加150mL丙酮一水溶液,密塞,搖勻,在20℃~25℃暗處浸泡48 h。24 h振搖一次,吸取10.0 mL上清液於25 mL具塞試管中,加2 g氯化鈉,密塞,劇烈振搖2 min,放置分層後,吸取5.O mL上清液於10 mL具塞試管中,加1 g尼水氯化鈣,密塞,劇烈振搖2 min,靜置30 min以上,供氣相色譜測定用。
氣相色譜參考條件
色譜柱=2m×3 mm(內徑)不鏽鋼柱,內裝塗有15%聚丙二醇(Poly propylene glycol)或15%Ucon LB-550X的60目~80目Chromosorb W。
溫度:柱溫:140℃;檢測器、進樣口溫度,200℃。
載氣:氮氣70 mL/min。
測定
每天做標準曲線。吸取O、0.5、1.0、3.O、5.O、7.O 二溴乙烷標準使用液(相當予O、0.1、0.2、0.6、1.O、1.4 ngEDB),直接進樣,與測得峰高繪製標準曲線。
準確吸取0.5 L試樣澄清液,進樣。為避免檢測器超載,可將樣液用無水丙酮稀釋10倍或100倍再進樣。每個樣液都進樣3次,取平均峰高與標準曲線比較定量。
結果計算
試樣中二溴乙烷的含量按式(7)進行計算。
X= A*1000/(m*V1/v2*1000) ……………………………………(6)
式中:
——試樣中二溴乙烷的含量,單位為微克每千克(mg/kg);
——進樣樣液相當二溴乙烷的質量,單位為納克();
——試樣進樣體積,單位為微升(mL);
——試樣質量,單位為克(g)。
125——150mL浸漬液中丙酮的體積,單位為毫升(mL)。
注:如無以上介紹的固定液,以下色譜柱也可用。
①20%OV-101塗於Chromosorb W HP 80目~100目上。
②30% DC-200塗於Gas Chrom Q 80目~100目上。
③10%DC-200塗予Chromosorb W AW DMCS 60目~80目上。
④6%OV-210+F4% SE-30塗於Gas Chrom Q 80目~100目上。
計算結果保留兩位有效數字。
