成分
化學名稱:
6-乙醯基-8-環戊基-5-甲基-2-[[5-(1-哌嗪基)-2-吡啶基]氨基]吡啶並[2,3-d]嘧啶-7(8H)-酮
化學式結構:
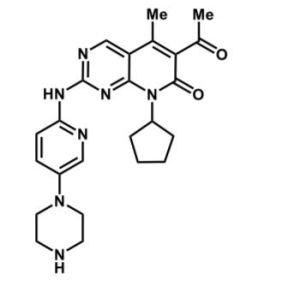
分子式:C24H29N7O2
分子量:447.54
性狀
本品為膠囊劑,內容物為類白色至黃色粉末。
適應症
本品適用於激素受體(HR)陽性、人表皮生長因子受體2(HER2)陰性的局部晚期或轉移性乳腺癌,應與芳香化酶抑制劑聯合使用作為絕經後女性患者的初始內分泌治療。
規格
(1)75mg;(2)100mg;(3)125mg
用法用量
應由具抗癌藥物使用經驗的醫生開始並監督本品治療。
推薦劑量
哌柏西利的推薦劑量為125mg,每天一次,連續服用21天,之後停藥7天(3/1給藥方案),28天為一個治療周期。治療應當持續進行,除非患者不再有臨床獲益或出現不可接受的毒性。
當與來曲唑聯用時,來曲唑的推薦劑量為2.5mg,口服,每天一次,在整個28天治療周期連續服藥。具體請參見來曲唑批准的說明書。
給藥方法
口服,應與食物同服,最好隨餐服藥以確保哌柏西利暴露量一致(見“藥代動力學”)。
哌柏西利不得與葡萄柚或葡萄柚汁同服(見“藥物相互作用”)。
哌柏西利膠囊應整粒吞服(吞服前不得咀嚼、壓碎或打開膠囊)。
如果膠囊出現破損、裂紋或其他不完整的情況,則不得服用。
應鼓勵患者在每天大約相同的時間服藥。
如果患者嘔吐或者漏服,當天不得補服。應照常進行下次服藥。
劑量調整
建議根據個體安全性和耐受性調整哌柏西利的劑量。出現某些不良反應時可能需要暫時中斷/延遲給藥和/或減低劑量,或永久停藥來進行控制,請參照表1、2和3中提供的方案進行劑量調整(見注意事項和不良反應)。

在開始哌柏西利治療前、每個治療周期開始時、前2個治療周期的第15天以及有臨床指征時應監測全血細胞計數。
對於前6個治療周期內發生最高嚴重程度為1或2級中性粒細胞減少症的患者,其後續周期的全血細胞計數監測時間應為每3個月一次、各周期開始之前以及有臨床指征時。
建議在中性粒細胞絕對計數(AbsoluteNeutrophilCount,ANC)≥1000/mm3且血小板計數≥50000/mm3時接受哌柏西利。
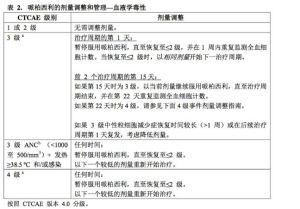
ANC=中性粒細胞絕對計數;
CTCAE=不良事件通用術語標準;LLN=正常值下限。
a表格適用於除淋巴細胞減少症以外的所有血液學不良事件(除非伴隨臨床事件,如機會性感染)
bANC:1級:ANC<LLN-1500/mm3;2級:ANC1000-<1500/mm3;3級:ANC500-<1000/mm3;4級:ANC<500/mm3。
表3.哌柏西利的劑量調整和管理—非血液學毒性

CTCAE級別1或2級:無需調整劑量。
CTCAE級別≥3級非血液學毒性(如果治療後仍然持續):暫停給藥,直至症狀緩解至:
≤1級;
≤2級(如果認為對患者的安全無風險)以下一個較低的劑量重新開始治療。
特殊人群
老年人
≥65歲的患者無需調整哌柏西利的劑量(見藥代動力學)。
兒科人群
尚未確定哌柏西利在≤18歲兒童和青少年患者中的安全性和療效。沒有數據可用。
肝損傷
輕度或中度肝損傷患者(Child-PughA級和B級)無需調整哌柏西利的劑量。重度肝損傷(Child-PughC級)患者的推薦劑量為75mg,每天一次,採用3/1給藥方案(見注意事項和藥代動力學)。
腎損傷
輕度、中度或重度腎損傷患者(肌酐清除率[CreatinineClearance,CrCl]≥15mL/min)無需調整哌柏西利的劑量。需要血液透析患者的數據不充分,無法對該人群提供任何劑量調整建議(見注意事項和藥代動力學)。
與CYP3A強效抑制劑合用時的劑量調整
避免伴隨使用CYP3A強效抑制劑,考慮替換為沒有或只有微弱CYP3A抑制作用的其他伴隨用藥。如果患者必須合用CYP3A強效抑制劑,則將哌柏西利的劑量減少至75mg,每天一次。如果停用強效抑制劑,則將哌柏西利的劑量增加至開始使用CYP3A強效抑制劑之前的劑量(在抑制劑的3至5個半衰期後)(參見藥物相互作用、藥代動力學和不良反應)。
不良反應
以下說明描述了在臨床試驗中觀察到的判斷為可能由哌柏西利引起的不良反應及其近似的發生率。由於每項臨床試驗的條件各不相同,在一個臨床試驗中觀察到的不良反應的發生率不能與另一個臨床試驗觀察到的不良反應發生率直接比較,也可能不能反映臨床實踐中的實際發生率。
安全性特徵
哌柏西利的總體安全性特徵評估來自在HR陽性、HER2陰性晚期或轉移性乳腺癌隨機研究中接受哌柏西利與內分泌療法聯合治療(527例與來曲唑聯用和345例與氟維司群聯用)的872例患者的合併數據[包括研究PALOMA-1(A5481003),研究PALOMA-2(A5481008),研究PALOMA-3(A5481023)]。
臨床研究中,接受哌柏西利治療的患者報告的最常見(≥20%)的任何級別的不良反應為中性粒細胞減少症、感染、白細胞減少症、疲乏、噁心、口腔炎、貧血、脫髮和腹瀉。哌柏西利的最常見(≥2%)的≥3級不良反應為中性粒細胞減少症、白細胞減少症、貧血、疲乏和感染。
在研究PALOMA-2中評估了哌柏西利(125mg/天)聯合來曲唑(2.5mg/天)治療對照安慰劑聯合來曲唑治療的安全性。哌柏西利聯合來曲唑的中位治療持續時間為19.8個月,而安慰劑聯合來曲唑的中位治療持續時間為13.8個月。在接受哌柏西利聯合來曲唑治療的患者
中,有36%的患者因任何級別的不良反應而減量。43/444(9.7%)例接受哌柏西利聯合來曲唑治療的患者以及13/222(5.9%)例接受安慰劑聯合來曲唑治療的患者發生了與不良反應相關的永久停藥。導致接受哌柏西利聯合來曲唑治療的患者永久停藥的不良反應包括中性粒細胞減少症(1.1%)和丙氨酸轉氨酶升高(0.7%)。
不良反應列表
表4報告了3隨機研究[研究PALOMA-1(A5481003),研究PALOMA-2(A5481008),研究PALOMA-3(A5481023)]的合併數據集中的不良反應。合併數據集中哌柏西利治療的中位持續時間為12.7個月。
表5報告了3項隨機研究的合併數據集中的實驗室檢查異常。
按系統器官分類和發生頻率列出不良反應。發生頻率定義為:十分常見(≥1/10)、常見(≥1/100至<1/10)和偶見(≥1/1,000至<1/100)。
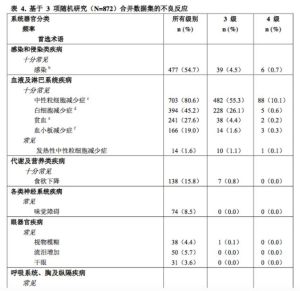
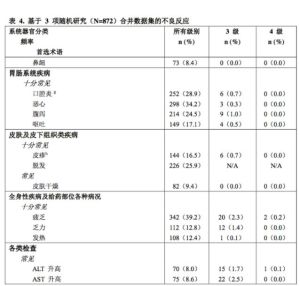
ALT=丙氨酸氨基轉移酶
AST=天冬氨酸氨基轉氨酶
N/n=患者人數
N/A=不適
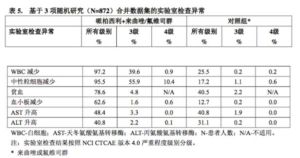
在PALOMA-2和PALOMA-3兩項研究中,納入了200例亞裔患者。在接受哌柏西利的亞裔患者中報告的3級或4級中性粒細胞減少症和白細胞減少症發生率高於非亞裔患者,因而在亞裔患者中的劑量中斷、劑量減少和周期延遲發生頻率也略高於非亞裔患者,但通過方案規定的劑量調整可控制總體安全性,亞裔患者與非亞裔患者具有相似的中位治療持續時間。根據對已有的哌柏西利劑量暴露、安全性和療效數據進行的累積分析,將125mg每日一次作為亞裔患者的起始劑量是恰當的。需根據患者個體的安全性和耐受性,嚴格遵循說明書調整哌柏西利劑量。
特定不良反應的描述
總體而言,3項隨機研究中有703例(80.6%)無論以何種聯用方案接受哌柏西利治療的患者報告了任何級別的中性粒細胞減少症,其中分別有482例(55.3%)和88例(10.1%)患者報告3級和4級中性粒細胞減少症(見表4)。
3項隨機臨床研究中,至首次發生任何級別中性粒細胞減少症的中位時間為15天(12-700天),≥3級中性粒細胞減少症的中位持續時間為7天。
0.9%接受哌柏西利與氟維司群聯用和2.1%接受哌柏西利與來曲唑聯用的患者報告了發熱性中性粒細胞減少症。
在總體臨床研究中,大約2%接受哌柏西利治療的患者曾報告過發熱性中性粒細胞減少症。
禁忌
對活性成分或章節“成分”項下所列的任一輔料過敏者禁用。
禁止使用含聖約翰草的製品(見藥代相互作用)。
注意事項
絕經前/圍絕經期女性
鑒於芳香化酶抑制劑的作用機制,絕經前/圍絕經期女性接受哌柏西利與芳香化酶抑制劑聯合治療時,必須進行卵巢切除或使用促黃體生成激素釋放激素(LuteinizingHormoneReleasingHormone,LHRH)激動劑抑制卵巢功能。哌柏西利聯合氟維司群用於絕經前/圍絕經期女性的研究中,僅與LHRH激動劑聯合用藥。
危重內臟疾病(轉移)
尚未在危重的有內臟疾病(轉移)患者中研究哌柏西利的療效和安全性(見【臨床試驗】)。
血液學毒性
中性粒細胞減少症是臨床研究中最常報告的不良反應,臨床研究中大約有2%的接受哌柏西利治療的患者曾報告過發熱性中性粒細胞減少症,並報告了1例中性粒細胞減少性敗血症引起的死亡。應在哌柏西利治療開始前、每個周期開始時、前兩個周期的第15天以及出現臨床指征時監測全血細胞計數。對於出現3或4級中性粒細胞減少症的患者,建議中斷給藥、減少劑量或延遲開始治療周期,並進行密切監測。(見用法用量和不良反應)。醫生應告知患者立即報告任何發熱事件。
感染
因為哌柏西利具有骨髓抑制特性,其可使患者易於出現感染。
多項隨機研究報導了哌柏西利組患者的感染率高於各自的對照組患者。分別有4.5%和0.7%的接受哌柏西利任何聯用方案治療的患者發生了3級和4級感染(見不良反應)。
應監測患者的感染體徵和症狀並且適當時應給予治療(見用法用量)。
患者在出現任何骨髓抑制或感染體徵或症狀時立即報告,例如發熱、寒戰、頭暈、氣短、無力或出血和/或瘀傷傾向加重。
肝損傷
中度或重度肝損傷患者應慎用哌柏西利,並密切監測毒性體徵(見用法用量和 藥代動力學)。
腎損傷
中度或重度腎損傷患者應慎用哌柏西利,並密切監測毒性體徵(見用法用量和 藥代動力學)。
與CYP3A4抑制劑或誘導劑聯合治療
強效CYP3A4抑制劑可導致毒性增加(見藥物相互作用)。哌柏西利治療期間應避免與強效CYP3A抑制劑合用。僅在認真評估潛在獲益和風險後才可考慮同時使用。如不能避免與強效CYP3A抑制劑同時使用,應將哌柏西利的劑量降至75mg每天一次。停止使用強效抑制劑時,應將哌柏西利的劑量(抑制劑的3-5個半衰期後)增加至開始使用強效CYP3A抑制劑前的劑量(見藥物相互作用)。
與CYP3A誘導劑同時使用可導致哌柏西利的暴露量降低,所以有缺乏療效的風險。因此,應避免哌柏西利與強效CYP3A4誘導劑合用。哌柏西利與中效CYP3A誘導劑同時使用時無需調整劑量(見藥物相互作用)。
有生育能力的女性或其配偶
有生育能力的女性或其男性配偶在使用哌柏西利治療期間必須使用一種高效的避孕方法(見孕婦及哺乳期婦女用藥)。
乳糖
哌柏西利含乳糖。存在半乳糖不耐症、Lapp乳糖酶缺乏症或葡萄糖-半乳糖吸收不良症等罕見遺傳疾病的患者不得服用哌柏西利。
對駕駛和操作機器能力的影響
哌柏西利對駕駛和操作機器能力的影響很小。但是,哌柏西利可能引起疲乏,患者在駕駛或操作機器時應謹慎。
孕婦及哺乳期婦女用藥
有生育能力的女性/避孕
接受本藥品治療的有生育能力的女性或其男性配偶,應在治療期間以及完成治療後分別至少3周(女性)或14周(男性)內採取充分的避孕措施(如,雙重屏障避孕)(見藥物相關作用)。
妊娠
尚缺乏關於孕婦使用哌柏西利的數據或數據有限。動物研究顯示哌柏西利具有生殖毒性(見藥理毒理)。不建議孕婦和未採取避孕措施的有生育能力的女性使用哌柏西利。
哺乳
尚未在人體或動物中進行相關研究以評價哌柏西利對乳汁生成的影響、是否存在於母乳中或對母乳餵養嬰兒的影響。尚不清楚哌柏西利是否會分泌至人類乳汁中。接受哌柏西利治療的患者不應哺乳。
生育力
在非臨床生殖毒性研究中,未發現對大鼠的發情周期(雌性)或交配和生育力(雄性和雌性)有影響。尚未獲得對人類生育力影響的臨床數據。根據非臨床安全性研究中雄性生殖器官的變化(睪丸曲細精管變性、附睪精子減少、精子活力和密度降低以及前列腺分泌減少),哌柏西利治療可能會損害男性的生育力(見藥理毒理)。因此,男性在開始哌柏西利治療前應考慮保存精液。
兒童用藥
尚未確定哌柏西利在18歲及以下的兒童和青少年患者中的安全性和療效。尚無相關數據。
老年用藥
在PALOMA-2研究中接受哌柏西利治療的444例患者中,181(41%)例患者≥65歲,48(11%)例患者≥75歲。未發現上述患者與年輕患者在哌柏西利的安全性或有效性方面存在差異。65歲及以上患者無需調整哌柏西利的劑量(見藥代動力學)。
藥物相互作用
哌柏西利主要被CYP3A和磺基轉移酶(Sulphotransferase,SULT)SULT2A1代謝。在體內,哌柏西利是CYP3A的時間-依賴性弱抑制劑。
其它藥品對哌柏西利藥代動力學的影響
CYP3A抑制劑的影響同時給予多劑量200mg伊曲康唑與單劑量125mg哌柏西利,相對於單獨給予單劑量125mg哌柏西利,哌柏西利的全身暴露量(AUCinf)和峰濃度(Cmax)分別增加了約87%和34%。
應避免與強效CYP3A抑制劑合用,包括但不限於:克拉黴素、茚地那韋、伊曲康唑、酮康唑、洛匹那韋/利托那韋、奈法唑酮、奈非那韋、泊沙康唑、沙奎那韋、特拉匹韋、泰利黴素、伏立康唑和葡萄柚或葡萄柚汁(見用法用量和注意事項)。
與輕度和中度CYP3A抑制劑合用時無需調整劑量。CYP3A誘導劑的影響同時給予多劑量600mg利福平與單劑量125mg哌柏西利,相對於單獨給予單劑量125mg哌柏西利,哌柏西利AUCinf和Cmax分別降低了約85%和70%。
應避免與強效CYP3A誘導劑合用,包括但不限於:卡馬西平、恩雜魯胺、苯妥英、利福平和聖約翰草(見禁忌和 注意事項)。
同時給予多劑量每天400mg莫達非尼(一種中效CYP3A誘導劑)與單劑量125mg哌柏西利,相對於單獨給予單劑量125mg哌柏西利,哌柏西利AUCinf和Cmax分別降低了約32%和11%。與中效CYP3A誘導劑合用時無需調整劑量(見注意事項)。
抗酸藥的影響
餐後(攝入中脂餐)同時給予多劑量質子泵抑制劑(ProtonPumpInhibitor,PPI)雷貝拉唑與單劑量125mg哌柏西利,相對於單獨給予單劑量125mg哌柏西利,哌柏西利Cmax降低了41%,但對AUCinf的影響有限(降低了13%)。
空腹條件下同時給予多劑量質子泵抑制劑(PPI)雷貝拉唑與單劑量125mg哌柏西利,哌柏西利AUCinf和Cmax分別降低了62%和80%。因此,哌柏西利應與食物同服,最好隨餐服用(見用法用量和 藥代動力學)。
鑒於H2受體拮抗劑和局部抗酸劑與PPI相比對胃內pH的影響較小,哌柏西利與食物同服時,預期H2受體拮抗劑或局部抗酸劑對哌柏西利的暴露量無臨床相關影響。
哌柏西利對其它藥品藥代動力學的影響
在每天給予125mg達到穩態後,哌柏西利是一種弱的時間-依賴性CYP3A抑制劑。與咪達唑侖單獨給藥相比,多劑量哌柏西利與咪達唑侖同時給藥時,咪達唑侖AUCinf和Cmax值分別增加了61%和37%。
治療指數狹窄的敏感CYP3A4底物(如阿芬太尼、環孢素、雙氫麥角胺、麥角胺、依維莫司、芬太尼、匹莫齊特、奎尼丁、西羅莫司和他克莫司)與哌柏西利同時使用時可能需要降低劑量,因為哌柏西利可增加它們的暴露量。
哌柏西利與來曲唑之間的藥物相互作用
一項乳腺癌患者臨床研究的藥物相互作用(Drug-DrugInteraction,DDI)評價部分的數據表明,哌柏西利與來曲唑聯用時,兩種藥品之間無藥物相互作用。
他莫昔芬對哌柏西利暴露量的影響
在健康男性受試者中進行的一項DDI研究的數據表明,單劑量哌柏西利與多劑量他莫昔芬同時給藥,與哌柏西利單獨給藥時的暴露量相當。
哌柏西利與氟維司群之間的藥物相互作用
在乳腺癌患者中進行的一項臨床研究的數據表明,哌柏西利與氟維司群聯用時,兩種藥品之間無臨床相關藥物相互作用。
哌柏西利與口服避孕藥之間的藥物相互作用
尚未對哌柏西利與口服避孕藥之間的DDI進行研究(見孕婦和哺乳期婦女用藥)。
與轉運蛋白的體外研究
根據體外研究數據,預計哌柏西利抑制腸道P-糖蛋白(P-Glycoprotein,P-gp)和乳腺癌耐藥蛋白質(BreastCancerResistanceProtein,BCRP)介導的轉運。因此,哌柏西利與P-gp(如地高辛、達比加群、秋水仙鹼)或BCRP(如,普伐他汀、瑞舒伐他汀、柳氮磺胺吡啶)的底物類藥品合併用藥可增加它們的治療作用和不良反應。
根據體外研究數據,哌柏西利可抑制攝取轉運體有機陽離子轉運蛋白OCT1,因此可增加該轉運蛋白的底物類藥品(如二甲雙胍)的暴露量。
藥物過量
尚無針對哌柏西利的特效解毒藥。如果哌柏西利用藥過量,可能出現胃腸道(如,噁心、嘔吐)和血液學(如,中性粒細胞減少症)毒性,應給予一般的支持性治療。
臨床試驗
隨機Ⅲ期研究PALOMA-2:哌柏西利與來曲唑聯用作為雌激素受體(ER)陽性、HER2陰性的晚期或轉移性乳腺癌患者初始內分泌治療。
在ER陽性、HER2陰性的不能通過手術切除或放療治癒的局部晚期乳腺癌患者或既往未接受過針對轉移灶的全身治療的晚期乳腺癌患者中進行了一項隨機、雙盲、安慰劑對照、國際多中心研究,評價了哌柏西利與來曲唑聯用和來曲唑與安慰劑聯用的療效。
總計666例絕經後婦女以2:1的比例隨機分配至哌柏西利+來曲唑組或安慰劑+來曲唑組,並按病灶部位(內臟、非內臟)、從完成(新)輔助治療至疾病復發的無病間期(新發轉移、12個月、>12個月)、既往(新)輔助抗腫瘤治療的類型(既往激素治療、無既往激素治療)分層。研究排除了存在晚期、症狀性、內臟轉移,短期內可能出現危及生命的併發症(包括大量積液無法控制[胸膜積液、心包液、腹膜積液]、肺淋巴管炎以及肝臟受累面積超過50%)的患者。
患者持續接受分配的治療,直到發生客觀疾病進展、症狀惡化、不可接受的毒性、死亡或撤銷同意書,以先發生者為準。不允許治療組間交叉治療。
哌柏西利+來曲唑組與安慰劑+來曲唑組之間患者的基線人口統計學以及預後特徵具有可比性。入組本研究的患者的中位年齡為62歲(範圍:28-89歲),多數患者為白種人(78%),且多數患者的美國東部腫瘤協作組(ECOG)體力狀態(PS)為0或1(98%)。在診斷為晚期乳腺癌前,48.3%患者接受過化療和56.3%患者接受過抗激素治療,37.2%的患者既往未接受過全身治療。大多數患者(97.4%)在基線時有轉移病灶,23.6%的患者只有骨轉移,49.2%的患者有內臟轉移。
研究的主要終點是由研究者按照實體瘤療效評價標準(ResponseEvaluationCriteriainSolidTumors,RECIST)v1.1評估的無進展生存期(Progression-FreeSurvival,PFS)。次要療效終點包括客觀緩解率(ObjectiveResponseRate,ORR)、臨床獲益緩解(ClinicalBenefitResponse,CBR)、安全性和生活質量(QualityofLife,QoL)變化。
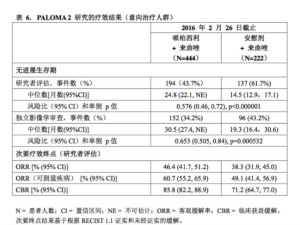
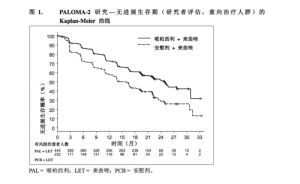
研究達到了主要終點。哌柏西利+來曲唑組與安慰劑+來曲唑組患者的中位PFS分別為24.8個月(95%CI:22.1,NE)和14.5個月(95%CI:12.9,17.1)。風險比(HazardRatio,HR)為0.576(95%CI:0.46,0.72),單側分層對數秩檢驗p值<0.000001。 PALOMA-2研究的療效數據總結於表6中,PFS的Kaplan-Meier曲線見圖1。
PALOMA-2研究的療效數據總結於表6中,PFS的Kaplan-Meier曲線見圖1。
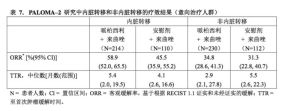
基於預後因素和極限特徵進行一些列預先定義的亞組PFS分析以考察治療效果的內部一致性。在所有個體患者亞組(按分層因素以及極限特徵定義)中觀察到了哌柏西利+來曲唑組可降低疾病進展風險或死亡風險。該結果在以下患者中較為顯著:內臟轉移患者(HR=0.67[95%CI:0.50,0.89],mPFS為19.2個月與12.9個月),或不伴內臟轉移的患者(HR=0.48[95%CI:0.34,0.67],mPFS為未達到[NR]與16.8個月),或僅發生骨轉移的患者(HR=0.36[95%CI:0.22,0.59],mPFS為NR與11.2個月),或沒有僅骨轉移的患者(HR=0.65[95%CI:0.51,0.84],mPFS為22.2個月與14.5個月)。與之相似,在512例通過免疫組化(Immunohistochemistry,IHC)檢測腫瘤Rb蛋白質表達結果呈陽性的患者中,觀察到哌柏西利+來曲唑的疾病進展或死亡風險下降(HR=0.531[95%CI:0.42,0.68],mPFS為24.2個月與13.7個月)。在51例通過IHC檢測腫瘤Rb蛋白質表達結果呈陰性的患者中,雖不具有統計學顯著性,但哌柏西利聯合來曲唑有利於疾病進展或死亡風險的下降(HR=0.675[95%CI:0.31,1.48],mPFS為NR與18.5個月)。
在伴或不伴內臟轉移的患者亞組中評估的其它療效指標(ORR和TTR)見表7。
藥理毒理
藥理作用
哌柏西利是細胞周期蛋白依賴性激酶(CDK)4和6的抑制劑。周期蛋白D1和CDK4/6位於細胞增殖信號通路的下游。在體外,通過阻滯細胞從G1期進入S期,而減少雌激素受體(ER)陽性乳腺癌細胞系的細胞增殖。哌柏西利和雌激素拮抗劑聯合作用於乳腺癌細胞系時,可降低視網膜母細胞瘤(Rb)蛋白磷酸化,從而導致E2F表達,及其信號傳導下降,與藥物各自單用相比具有更強的生長抑制作用。哌柏西利和雌激素拮抗劑聯合作用於ER陽性的乳腺癌細胞系時,與藥物各自單用相比,可使細胞老化增加,這一效應在哌柏西利停藥後最多維持6天,但抗雌激素治療繼續進行時,可導致更大程度的細胞老化。人源性ER陽性乳腺癌異種移植模型體內研究顯示,與藥物各自單用相比,哌柏西利與來曲唑聯用可對Rb磷酸化、下游信號傳導以及腫瘤生長產生更強的抑制作用。
人骨髓單核細胞體外給予哌柏西利,無論有無抗雌激素處理,未見細胞發生老化,去除哌柏西利後細胞可恢復增殖。
毒理研究
一般毒性
在犬遙測試驗中,給藥劑量在人體臨床暴露量(Cmax)4倍以上時,可見心血管影響(QTc延長、心率下降、RR間期延長和收縮壓升高)。
在一項大鼠27周重複給藥毒性試驗中,大鼠在試驗早期尚未成熟,發現與胰腺(胰島細胞空泡形成)、眼睛(白內障、晶狀體變性)、腎臟(腎小管空泡形成、慢性進行性腎病)和脂肪組織(萎縮)變化相關的葡萄糖代謝改變(尿糖、高血糖症、胰島素下降),這種現象在哌柏西利經口給藥劑量≥30mg/kg/天(AUC約為臨床推薦劑量下的成人人體暴露量的11倍)的雄鼠中發生率最高。其中一些不良反應(尿糖/高血糖症、胰島細胞空泡形成和腎小管空泡形成)在未成熟大鼠中進行的15周重複給藥毒性試驗中發生率和嚴重程度較低。在試驗開始時已成熟的大鼠中進行的27周重複給藥毒性試驗,以及39周犬重複給藥毒性試驗中未見葡萄糖代謝改變或胰腺、眼睛、腎臟和脂肪組織相關變化。在大鼠中可見與葡萄糖代謝改變無關的牙齒毒性。哌柏西利以100mg/kg給藥劑量給藥27周(AUC約為臨床推薦劑量下的成人人體暴露量的15倍)可導致大鼠切牙生長異常(變色、造釉細胞變性/壞疽、單核細胞浸潤)。
遺傳毒性
哌柏西利Ames試驗和體外人淋巴細胞染色體畸變試驗結果陰性,中國倉鼠卵巢細胞體外試驗、雄性大鼠骨髓試驗微核試驗結果陽性。
生殖毒性
在雌性大鼠生育力試驗中,給藥劑量高達300mg/kg/天(AUC約為人體臨床暴露量的4倍)時,未見哌柏西利對動物交配或生育力產生影響。在大鼠、犬重複給藥毒性試驗中,大鼠給藥劑量高達300mg/kg/天,犬給藥劑量高達3mg/kg/天(AUC分別約為臨床推薦劑量下人體暴露量的6倍以及與人體暴露量相當)時,未見哌柏西利對雌性動物生殖器官產生任何不良影響。在大鼠和犬重複給藥毒性試驗、以及大鼠雄性生育力試驗中可見哌柏西利對雄性生殖系統和生育力產生不良影響。重複給藥毒性試驗中,大鼠和犬分別給予哌柏西利≥30mg/kg/天和≥0.2mg/kg/天(AUC分別約為臨床推薦劑量下人體暴露量的≥10倍和≥0.1倍)時,可見給藥相關的睪丸、附睪、前列腺和精囊器官重量下降、萎縮或變性、精子減少、小管內細胞碎片和分泌減少,分別經過4周和12周的停藥期後,上述對大鼠和犬雄性生殖器官的影響部分可逆。在雄性大鼠生育力和早期胚胎髮育毒性試驗中,哌柏西利給藥劑量為100mg/kg/天(推算AUC約為臨床推薦劑量下人體暴露量的20倍)時未見對交配產生影響,但生育力出現輕微下降,表現為精子活力和密度較低。在雌性大鼠生育力和早期胚胎髮育毒性試驗中,從交配前15天至妊娠第7天經口給予哌柏西利,在劑量達到300mg/kg/天(母體全身暴露約為臨床推薦劑量下的人體暴露量的4倍)時未見導致胚胎毒性。
在大鼠和兔胚胎-胎仔發育試驗中,妊娠動物在器官形成期分別經口給予哌柏西利高達300mg/kg/天和20mg/kg/天,大鼠在母體毒性劑量300mg/kg/天時可引起胎仔毒性,導致胎仔體重下降,劑量≥100mg/kg/天時,骨骼變異的發生率增加(第七頸椎出現肋骨的發生率增加)。兔在母體毒性劑量20mg/kg/天時,骨骼變異(包括前肢小趾骨)的發生率增加。大鼠劑量為300mg/kg/天和兔劑量為20mg/kg/天時,母體全身暴露量(AUC)分別約為臨床推薦劑量下人體暴露量的4倍和9倍。
文獻報導,CDK4/6雙基因敲除小鼠在胎仔發育晚期(妊娠第14.5天至出生)因重度貧血死亡。但是由於靶點抑制程度存在差異,基因敲除小鼠數據可能無法預測對人的影響。
致癌性
尚未進行致癌性試驗。
藥代動力學
在實體瘤患者(包括晚期乳腺癌)和健康志願者中研究了哌柏西利的藥代動力學特徵。
吸收
哌柏西利一般在口服後6-12小時之間達峰濃度(Cmax)。口服125mg哌柏西利後,其平均絕對生物利用度為46%。在25-225mg劑量範圍時,血藥濃度時間曲線下面積(AreaUndertheCurve,AUC)和Cmax通常隨劑量成比例增加。在每天一次重複給藥後8天內達到穩態。哌柏西利按每天一次重複給藥可出現蓄積,中位蓄積比為2.4(範圍:1.5-4.2)。
食物影響
在大約13%的空腹人群中,哌柏西利的吸收和暴露量極低。在這一小部分人群中,進食增加了哌柏西利的暴露量,但在其餘人群中,進食對哌柏西利的暴露量沒有臨床相關影響。與禁食過夜後給藥相比,哌柏西利與高脂食物同服時AUCinf和Cmax分別升高了21%和38%,與低脂食物同服時分別升高了12%和27%,而在哌柏西利給藥前1小時和給藥後2小時進食中脂食物時分別升高了13%和24%。此外,進食還顯著降低了個體間和個體自身的哌柏西利暴露量差異。根據上述結果,哌柏西利應與食物同服(見用法用量)。
分布
哌柏西利在體外與人血漿蛋白的結合率為85%,無濃度依賴性。在體內,人體血漿中哌柏西利的平均游離分數(fu)隨肝功能惡化程度逐漸增加。在體內,隨腎功能惡化,人體血漿中哌柏西利的平均fu無明顯變化趨勢。在體外,人體肝細胞主要通過被動擴散攝取哌柏西利。哌柏西利不是OATP1B1或OATP1B3的底物。
生物轉化
體外和體內研究表明哌柏西利經由肝細胞進行廣泛代謝。人單次口服[14C]標記的哌柏西利125mg後,哌柏西利的主要代謝途徑是磺化和氧化,次要途徑是葡萄糖苷酸化和醯化。血循環中檢測到的主要為哌柏西利原型藥。
大部分以代謝物形式排泄。哌柏西利的氨基磺酸結合物是在糞便中發現的主要藥物相關成分,占給藥劑量的25.8%。採用人肝細胞、肝胞漿和人肝S9組份以及重組磺基轉移酶(SULT)酶進行的體外研究表明主要參與哌柏西利代謝的酶為CYP3A和SULT2A1。
消除
在晚期乳腺癌患者中,哌柏西利的幾何平均表觀口服清除率(CL/F)為63L/h,平均血漿消除半衰期為28.8小時。6例健康男性受試者單次口服[14C]哌柏西利後,在15天內回收到了總放射量的92%(中位數);糞便(劑量的74%)為主要排泄途徑,尿中回收了17%的劑量。經糞便和尿液排泄的哌柏西利原型藥的回收率分別占給藥劑量的2%和7%。
在體外研究中,在臨床相關濃度時,哌柏西利不是CYP1A2、2A6、2B6、2C8、2C9、2C19和2D6的抑制劑,也不是CYP1A2、2B6、2C8和3A4的誘導劑。
體外評價表明,在臨床相關濃度時,哌柏西利對有機陰離子轉運體(OrganicAnionTransporter,OAT)1、OAT3、有機陽離子轉運體(OrganicCationTransporter,OCT)2、有機陰離子轉運多肽(OrganicAnionTransportingPolypeptide,OATP)1B1、OATP1B3和膽鹽輸出泵(BileSaltExportPump,BSEP)活性的抑制作用較弱。
特殊人群
年齡、性別和體重
基於一項包括183例癌症患者(50例男性和133例女性患者,年齡範圍:22-89歲,體重範圍:38-123kg)的群體藥代動力學分析,性別對哌柏西利的暴露量沒有影響,年齡和體重對哌柏西利的暴露量沒有具臨床意義的影響。
兒科人群
尚未在年齡≤18歲的患者中評估哌柏西利的藥代動力學。
肝損傷
在不同程度肝功能受試者中進行了一項藥代動力學試驗,數據表明,與肝功能正常受試者相比,輕度肝損傷(Child-PughA級)受試者中游離的哌柏西利暴露量(游離AUCinf)降低17%,而中度(Child-PughB級)和重度(Child-PughC級)肝損傷受試者分別增加34%和77%;輕度、中度和重度肝損傷受試者中游離的哌柏西利峰濃度(Cmax)分別增加7%、38%和72%。此外,基於一項包括183例晚期癌症患者的群體藥代動力學分析,其中包括40例輕度肝損傷患者(基於NCI分類;總膽紅素≤ULN和AST>ULN,或總膽紅素>1.0-1.5×ULN和任意水平AST),輕度肝損傷對哌柏西利的藥代動力學無影響。
腎損傷
在不同程度腎功能受試者中進行了一項藥代動力學試驗,數據表明,與腎功能正常(CrCl≥90mL/min)受試者相比,輕度(60mL/min≤CrCl<90mL/min)、中度(30mL/min≤CrCl<60mL/min)和重度(CrCl<30mL/min)腎損傷受試者對哌柏西利的總暴露量(AUCinf)分別增加39%、42%和31%;哌柏西利峰暴露量(Cmax)分別增加17%、12%和15%。此外,基於一項包括183例晚期癌症患者的群體藥代動力學分析,其中包括73例輕度腎損傷患者和29例中度腎損傷,輕度和中度腎損傷對哌柏西利的藥代動力學無影響。尚未在需要血液透析患者中研究哌柏西利的藥代動力學。
亞裔人群
在日本健康志願者中進行了一項藥代動力學試驗,與非亞裔受試者相比,日本受試者單次口服給藥後的哌柏西利AUCinf和Cmax分別高出30%和35%。但在後續研究中,日本或亞裔乳腺癌患者接受多次給藥後未觀察到上述結果。基於亞裔和非亞裔人群的累積藥代動力學、安全性和療效數據分析,不需要基於亞裔人種進行劑量調整。
中國人群
研究A5481019(n=26)在既往未接受過任何針對晚期疾病的全身性抗癌治療的ER陽性、HER2陰性的絕經後晚期乳腺癌中國患者中評價哌柏西利與來曲唑聯合治療的PK特徵。該研究中所觀測到的中國患者哌柏西利的藥代動力學特徵與PALOMA-2和PALOMA-3研究中非中國患者的藥代動力學特徵一致。在A5481019研究中的中國患者的谷濃度與PALOMA-2研究中所觀測到的谷濃度一致,不需要基於中國人群進行劑量調整。
貯藏
室溫保存。開封后的藥品請保存於原包裝瓶內。
包裝
帶聚丙烯(PP)蓋的高密度聚乙烯(HDPE)瓶包裝,每瓶含有21粒膠囊。
有效期
36個月。
