病因
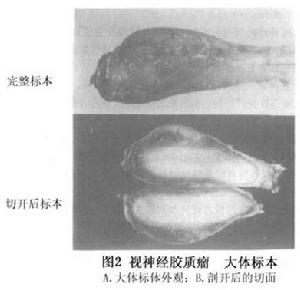 圖2 視神經膠質瘤
圖2 視神經膠質瘤發病機制
發病機制並不十分清楚。
流行病學
膠質瘤是顱內腫瘤中最常見的腫瘤 約占50%,而發生於視神經者並不多見,只占神經系膠質瘤的1%~2%。有人在1991年對1422例眼眶腫瘤進行病理分析,視神經膠質瘤18例,占1.3%。大批病例統計資料說明視神經膠質瘤占眶內腫瘤的1%~6%。
視神經膠質瘤多發於學齡前兒童,10歲以內占75%,90%見於20歲以前。文獻報告發現症狀最大年齡79歲。發病年齡與腫瘤部位有一定關係,發生於視交叉的視神經膠質瘤年齡較大。有明顯的性別傾向,發生於視神經者男女之比為1∶1~1∶3。但發生於視交叉時男女幾乎相等。
臨床表現
視神經膠質瘤的臨床表現與原發位置有明顯關係,一般均有視力減退、眼球突出和視盤水腫或萎縮,其他如斜視、眶深部腫物、眼球運動障礙和皮膚棕色素斑也常發生。膠質細胞位於視神經內部,該細胞的瘤性增殖,首先壓迫視神經纖維,引起視力減退和視野缺失。95%病例視力減退為第一症狀,60%以上患者就診時視力低於0.1,特別是原發於視神經管內的,視力喪失更為嚴重。腫瘤波及視交叉,影響雙眼視力及視野。腫瘤壓迫瞳孔反射纖維,引起上行性瞳孔對光反應遲鈍。單眼視力喪失,可發生廢用性斜視,引起家長注意而求醫 兒童雙眼視力喪失,可發生眼球震顫。一側性眼球突出也是視神經膠質瘤的常見體徵,原發於眶內段視神經的腫瘤均有這一體徵。眼球突出為無痛性漸進性,突出程度突然增加,並伴有視力喪失可能是腫瘤內囊樣變、囊內液
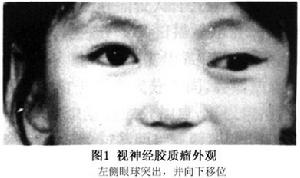 圖1 視神經膠質瘤
圖1 視神經膠質瘤增多或囊內出血所致。眼球突出程度一般為輕度或中度。由於視神經位於肌肉圓錐內, 視神經神經多為軸性眼球突出,腫瘤較大時眼球突出方向也可偏離軸位, 且多向外下方移位(圖1)。眼底檢查可發現視盤的改變 視盤水腫或萎縮。在大批量病人中 二者發生率幾乎相等。腫瘤位於視神經前段,影響軸漿流動,多發生視盤水腫。原發位置在視神經後段,特別是管內或顱內段 視盤原發萎縮多見。Wright發現病情活動時視盤水腫多見 病情穩定後視盤萎縮多見。在少數病例中可見腫瘤向前發展至視盤,眼底鏡下可見灰色腫物,突入玻璃體腔內。視盤表面偶見視神經睫狀靜脈,這種血管是視網膜中央靜脈與脈絡膜靜脈之間的側支循環, 眼球運動障礙較為少見,多發生於晚期。腫瘤體積較大時,偏心增長,有時可於眶緣捫及腫物。
兒童視神經膠質瘤與神經纖維瘤病有密切關係, 在膠質瘤病例中,有1/5~1/2可見虹膜淡黃色結節、皮膚咖啡樣色素斑、皮下軟性腫物及眶骨先天性缺失等神經纖維瘤病體徵。存在這些體徵而伴有視力喪失和原發性視神經萎縮時,應高度懷疑後部視神經膠質瘤的可能。神經纖維瘤病的存在,不影響膠質瘤的病程和預後。
併發症: 視神經膠質瘤顱內蔓延至視交叉可引起頭痛、 嘔吐、 癲癇及昏迷。
診斷
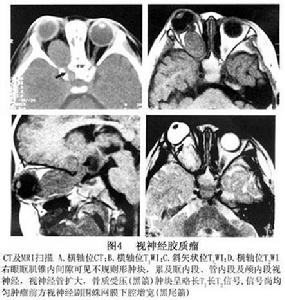 圖4 視神經膠質瘤
圖4 視神經膠質瘤鑑別診斷
影像學鑑別診斷:①視神經腦膜瘤:好發於成年女性,視力障礙,多在眼球突出之後 視神經鞘腦膜瘤可為偏心性,CT值較高,可伴有斑點、環形或不規則的鈣化,MRI 檢查T1WI、T2WI均呈中等信號,CT或MRI增強後可見“軌道征”;②視神經炎:臨床上表現視力急劇下降,可伴有眼球轉動時疼痛和眼眶深部脹痛等症狀。MRI表現為視神經瀰漫性增粗 一般不形成軟組織腫塊,T1WI視神經信號減低,T2WI信號增高,STIR呈高信號,增強後可有強化,以增強掃描聯合脂肪抑制序列顯示最佳, 可為多發性硬化的一種改變,MRI顯示腦室周圍硬化斑塊,則可確定本病。
另外,主要和一些能夠引起視神經增粗的疾病區別,如:炎性假瘤、腦膜瘤等。炎性假瘤常有炎症的表現,且視神經增粗的形狀常呈不規則增粗,所以與膠質瘤容易鑑別。腦膜瘤多見於成年人,視神經增粗的形狀各異,邊界不規則, 臨床鑑別有困難時應進行活檢證實診斷。
病理學檢查
正常星形膠質細胞分為纖維和原漿兩型,這兩型細胞均可發生腫瘤。兒童時期視神經膠質瘤幾乎均屬於纖維星形膠質細胞瘤, 根據細胞分化程度,纖維型星形膠質細胞瘤又分為4級:Ⅰ、Ⅱ級為良性,Ⅲ、Ⅳ級為惡性。兒童時期視神經纖維型星形膠質細胞瘤多為Ⅰ級,成年視神經星形膠質細胞瘤可見Ⅱ級,均屬於良性腫瘤。
視神經星形膠質細胞瘤大體病理標本檢查可見視神經呈梭形腫大(圖2),最大橫徑可達2.5cm,表面光滑,硬腦膜完整被撐大,淡白色,鮮嫩,類似於半透明。腫瘤沿視神經縱軸蔓延,常在視神經管內段變細。或單純的視神經和視束增粗,視交叉增寬。少數病例視交叉和視束增寬限於患側 腫瘤橫切面可見外為增厚的腦膜,內為灰白色細膩脆軟的腫瘤實質 以刮匙很易切割,吸引器吸除。由於瘤細胞增生,使小血管阻塞,影響神經纖維的營養,約有1/3的標本可見囊樣變,囊內充滿透明漿液和黏液體,Alcian藍呈陽性反應。嚴重囊樣變性者,可表現為囊性腫物,僅囊壁殘餘少量瘤細胞組織。
鏡下所見瘤細胞浸潤性擴大,與正常視神經纖維間缺乏明顯邊界。腫瘤由分化很好的星形膠質細胞構成, 瘤細胞細長,有頭髮樣突起,平行或編織狀排列(圖3)。軟腦膜結締組織隔增厚失去原有的結構,被瘤細胞擴大分開。在瘤細胞之間,散在少數正常的少突膠質細胞。磷鎢酸蘇木精(phosphotungstic acidic hematoxylin PTAH)染色瘤細胞星狀突起呈陽性。免疫組織化學染色,膠原纖維酸性蛋白質(glial fibrllary acid protein,GFAP)和神經元特殊磷鎢酸丙酮酸水合酶呈陽性。在細胞突內有嗜伊紅Rosenthal小體,PTAH呈強陽性。在腫瘤表面,可見蛛網膜細胞明顯增生,腦膜增厚, 有時誤診為腦膜瘤。
電鏡觀察,瘤細胞顯示纖維型星形膠質特徵。星形突起內充滿細絲狀物,直徑50~100nm和無定形的物質融合在一起,這便是光鏡所見的Rosenthal小體。Rosenthal小體為該腫瘤特徵發現, 星形膠質瘤Ⅱ級,瘤細胞較多,排列較密,形狀不整齊,原漿突起較粗,也稱星形母細胞瘤, 仍屬於良性範圍。
其它輔助檢查
1.X線檢查 腫瘤較小時,常無陽性改變。較大的腫瘤引起視神經孔向心性擴大,但骨皮質邊緣清楚,管壁一般不出現骨質硬化或破壞。如果同一病人兩側視神經孔大小相差超過1mm或單側視神經孔寬度超過5mm都要考慮異常,累及視交叉時在頭顱側位片上蝶鞍可呈“梨狀” 、“葫蘆狀”或擴大。
2.超音波探查
(1)B型超聲:顯示視神經梭形或橢圓性腫大,邊界清楚銳利。內回聲缺乏、少或中等。軸位掃描腫瘤後界不能顯示,探頭傾斜可顯示腫瘤後界呈中等回聲, 合併視盤水腫者,腫物回聲與隆起的視盤前強回聲光斑相連。眼球轉動時腫瘤前端反方向運動, 說明腫瘤與眼球關係密切,還可見眼球後部受壓變平。
(2)CDI:在腫瘤周邊可見血流,但不豐富。
 圖5 視神經膠質瘤
圖5 視神經膠質瘤4.MRI 表現為視神經呈梭形、冠狀或橢圓形增粗,多數為中心性,少數為偏心形。與正常眼外肌比較,視神經膠質瘤在T1WI呈低信號,T2WI呈高信號 增強後中度強化(圖5)。部分腫瘤壓迫使其前部正常的蛛網膜下腔擴大,表現為與腦脊液信號相似的長T1、長T2信號(圖6);由於少數腫瘤周圍蛛網膜等結構反應性增生而形成假性包膜,表現為長T1、長T2 。MRI可清楚顯示視神經膠質瘤的形態及其與鄰近結構的關係,也可清楚準確顯示視神經管內視神經膠質瘤,更直觀顯示視交叉或視束膠質瘤的形態及其侵犯的結構,如下丘腦、顳葉等,以增強掃描聯合脂肪抑制技術顯示最佳。
治療及預後
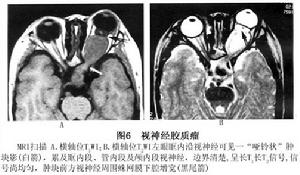 圖6 視神經膠質瘤
圖6 視神經膠質瘤不少學者認為兒童視神經膠質瘤是一種良性錯構瘤,發展甚慢,或到一定程度便停止進展 在視力良好情況下,活檢得到組織學證據後,可以在臨床密切觀察 不急於其他治療 另外一些學者則持截然相反的意見,認為多數病例最終將蔓延至視交叉 視束,影響兩側視力,繼續發展侵犯第三腦室及腦幹, 顱壓增高甚至死亡 不宜消極觀察。放射治療對視神經膠質瘤雖有一定效果,也只有15%~39%病例視力改進。但放療後視力仍可惡化,表明病變進展,所以多數病例仍需手術治療。Anderson等認為即便手術不能完全切除,去除腫瘤中大部分,消除了對周邊細胞的刺激,部分腫瘤切除也不再增長。Wright根據腫瘤的發展趨勢和視力情況確定治療方案是可取的。通過臨床和影像觀察把視神經膠質瘤分為穩定組與活動組 在穩定組初診時雖然視力也有減退、眼球突出、視盤水腫或萎縮,CT發現患例視神經腫大 甚至對側視神經和視交叉加寬,但在觀察過程中視力無明顯減退 眼球突出度增加緩慢,影像學顯示腫瘤無明顯增大者,則採取定期檢查,以便保留較好的視力。如在觀察中發現視力不斷減退, 眼球突出漸增進,超聲、CT和MRI發現腫瘤進展,應儘早手術切除。
關於手術進路,腫瘤僅限於眶內,外側開眶即可完全切除。前至眼球後極,後至眶尖、全眶內段視神經及腫瘤全部切除。如增粗的視神經已達到眼球部,視神經在眼球斷端電凝破壞。
X線和CT發現視神經孔(管)擴大並不是外側開眶的禁忌, 膠質瘤繼發腦膜細胞增生可以引起此種改變。MRI發現管內、顱內視神經侵犯或在觀察過程發現臨床症狀、體徵和影像顯示進展明顯, 用外側開眶或經顱手術, 自視交叉切除全部視神經,術後放療,多數病例長期觀察病變均無明顯發展。
雙側視神經膠質瘤,視交叉或視束腫瘤 經顱手術也難以完全切除, 保持視力和生命均有困難,在此情況只能採用放射治療。利用60Co或加速器照射病灶每4~6周40~60Gy。曾有報告視神經膠質瘤侵犯視交叉10例, 放射治療,隨訪0.5~17年無一例死亡, 且視力有所改進。對於手術切除有困難者,立體定位γ刀治療也有效。如腫瘤影響腦脊液循環,顱內壓增高, 可採用神經外科減壓,以減少痛苦延長生命。
對於視神經膠質瘤的治療意見比較混亂,利用臨床資料、超聲和CT肯定診斷,MRI確定病變範圍後可採用以下方案:①保留著有用的視力,眼球突出不明顯,MRI(強化T1WI和脂肪抑制)發現腫瘤距視神經管較遠,則定期觀察;②視力少於指數,眼球突出明顯,影響外觀,腫瘤限於眶內或觀察過程中腫瘤進展,外側開眶,手術切除, 切除斷端仍有瘤細胞者60Co側野照射40Gy或γ刀治療;③MRI顯示腫瘤已侵犯視神經管,顱內視神經和(或)患側視交叉,經顱開眶,切除視交叉至眼球後極部的視神經和腫瘤,視交叉端有瘤細胞者,放射治療40Gy或γ刀治療;④廣泛的視交叉部位侵犯或雙側視神經膠質瘤,放射治療。
預後
視神經膠質瘤是一種良性病,預後較好。少數病例有自發消退傾向,有的對放射治療有反應。手術治療未完全切除,往往也不再增長。但在年幼患兒術後可繼續擴大,且手術年齡越小,繼續增長可能性越大。
