簡介
神經元腫瘤和神經元與神經膠質混合性腫瘤,英文名:neuronal tumor & neuroglial and neuronal mixed tumor 。顱內神經元和混合性神經元腫瘤是少見的顱內腫瘤。病史較長,可1~5 年,表現為頭痛、癲癇發作等一般症狀,局限性定位體徵不常見,少數位於功能區而有相應表現。位於下丘腦的腫瘤可出現腦積水與下丘腦損害表現,如垂體功能低下、早熟、飲食亢進、嗜睡、指端肥大及糖尿病等。概述
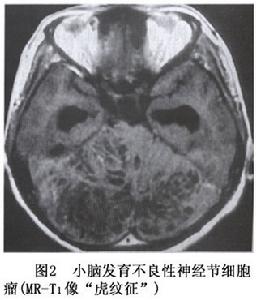 圖2
圖21.小神經元細胞腫瘤 有中央神經細胞瘤與嗅神經母細胞瘤。
2.大神經元細胞腫瘤 有神經節細胞瘤、Lhermitte-Duclos病等。
3.神經元與神經膠質混合性腫瘤 如神經節膠質瘤與間變性神經節膠質瘤。
4.松果體腺細胞腫瘤 雖已將其歸為單獨一類但也屬於神經元細胞腫瘤。
神經節膠質瘤是中樞神經系統少見的腫瘤,由Courville在1930年首先發現並命名這種瘤起源於胚胎未分化細胞,最終分化為成熟的神經元細胞與膠質細胞。因腫瘤生長緩慢以前曾一度將其歸為錯構瘤目前絕大多數作者認為這是一種有神經膠質瘤細胞與神經元瘤細胞的良性混合性腫瘤。
小腦發育不良性神經節細胞瘤(LDD)為小腦神經節細胞過度增生取代顆粒細胞與浦肯野細胞形成的錯構瘤樣病變至2003年文獻上共報導140例。本病由Lhermittle和Duclos在1920年報導,文獻上常稱之為LDD也稱之為小腦錯構瘤、浦肯野瘤或小腦良性肥大。本病可發生在任何年齡,以中青年居多,平均年齡為29歲和34歲,病程長,進展緩慢,主要症狀為顱壓增高、後組腦神經麻痹和小腦損害征。
胚胎髮育不良性神經上皮腫瘤(DNT)是1993年WHO中樞神經系統腫瘤分類中新加入的一種神經元和神經膠質細胞來源的腫瘤,這是一種在對癲癇患者行癲癇灶切除後對其進行組織學檢查所發現的一種良性腫瘤1988年Daumas-Duport等首次報導了39例並對其進行了詳細討論,所有腫瘤均位於皮質內,由星形細胞、少枝膠質細胞和神經元構成。
流行病學
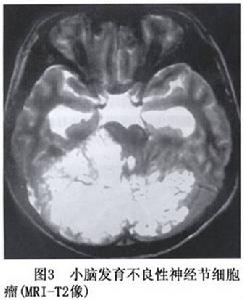 圖3
圖3LDD極少見,文獻報導的僅71例,其中有11例伴發Cowden綜合徵但最近Robinson等(2000)回顧過去40年中所見的5例LDD,都有Cowden病的表現,其中3例當時未曾診斷,說明過去的診斷有極大的不足。在國內研究近25年的14803例中樞神經系統腫瘤手術標本中病理證實的LDD只有1例。發生於一男性青年
DNT少見,自Daumas-Duport首批報導39例以來,以後均只有零星個例報導腫瘤多見於兒童,但也有青年患者,男女性無明顯差異。DNT好發於幕上,62%~78%位於顳葉,其餘幾乎均位於額葉。有人報導DNT占20歲以下組神經上皮性腫瘤的1.2%全年齡組的0.63%
病因
LDD病因尚未完全明確,Yachnis認為是小腦發育不良所引起近來發現部分LDD患者有家族史,不少患者合併有Cowden綜合徵(全身黏膜、皮膚多發性錯構瘤與腫瘤,包括腸息肉病、甲狀腺腫、乳腺纖維囊性病、乳腺癌及甲狀腺癌等)。分子生物學研究發現多數LDD腫瘤細胞中的10號染色體上的PTEN/MMAC1抑癌基因的5號外顯子有缺失這恰好與Cowden綜合徵病人中的發現相類同為此已有人提出本病實際上為斑痣性錯構瘤病的一種類型。
胚胎期發育不良性神經上皮腫瘤由多種神經細胞組成並伴有皮質發育不良,因此認為DNT為一種胚胎期發育不良而形成的腫瘤另有作者提出DNT事實上是由排列異位、紊亂的正常神經元與神經膠質細胞構成的錯構瘤。
發病機制
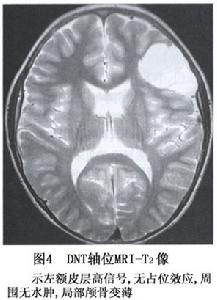 圖4
圖4小腦發育不良性神經節細胞瘤大體上為小腦腦葉增大、肥厚變形,腫瘤邊界欠清,表面呈黃白色,質地硬,血供不豐富。鏡下見小腦半球白質減少,小腦顆粒層有異常肥大的神經節細胞,顆粒細胞與浦肯野細胞明顯減少分子層增寬含有大量有髓神經纖維,肥大的神經節細胞的軸突過度髓鞘化且在軟膜下腔走行腦白質萎縮。增生的神經節細胞的軸突朝著皮質方向平行的排列少數細胞有核分裂。免疫組化染色發現在神經節細胞內突觸素為強陽性,而維蒙亭(vimentin)為陰性。
胚胎髮育不良性神經上皮腫瘤位於皮質結節狀,瘤組織鬆散,瘤內部分為囊性。腫瘤由神經元、少突細胞和星形細胞構成,腫瘤周圍皮質有局灶性發育不良神經元排列紊亂,瘤周白質內可見移位神經細胞。在低級別的膠質瘤區域間分布有小神經元區及大神經節細胞區,從而形成多結節樣改變瘤內囊腔內含有酸性黏液。部分毛細血管周圍由膠質纖維形成鞘,四周繞有小而圓的少枝細胞其中有時可見有神經元。腫瘤內無壞死、血管內皮增生及瘤細胞核分裂。免疫組化染色可見瘤內細胞密集區MIB-1染色陽性,血管周膠質鞘GFAP陽性。
臨床表現
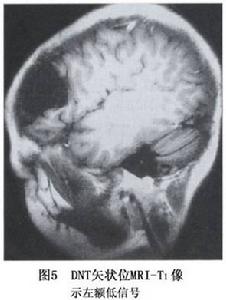 圖5
圖5小腦發育不良性神經節細胞瘤發生在小腦,有一定的占位效應,可梗阻腦脊液循環而致幕上腦室擴大第四腦室輕度移位,損害小腦臨床上主要以顱高壓症狀與腦積水為主要表現,後期可有小腦症狀與腦神經受損表現。約1/3患者可有巨顱症。伴有Cowden綜合徵者另可伴發全身皮膚黏膜上的錯構瘤及其他部位的腫瘤或腫瘤樣病變。少數病人可有患側後組腦神經麻痹等。
胚胎髮育不良性神經上皮腫瘤病程長,常在幼年或年輕時發病,主要表現為複雜性的局灶性癲癇發作。癲癇常為頑固性而不易控制。腦電圖常有病灶部位的癲癇波存在。因本病生長緩慢,局部顱骨可受壓變薄。
併發症
如進行手術治療,可能發生以下併發症:
1.顱內出血或血腫 與術中止血不仔細有關,隨著手術技巧的提高,此併發症已較少發生。創面仔細止血關顱前反覆沖洗,即可減少或避免術後顱內出血
2.腦水腫及術後高顱壓 可用脫水藥物降低顱內壓糖皮質激素減輕腦水腫
3.神經功能缺失 與術中損傷重要功能區及重要結構有關,術中儘可能避免損傷,出現後對症處理。
診斷
根據上述臨床表現及輔助檢查,一般可以做出診斷。
檢查
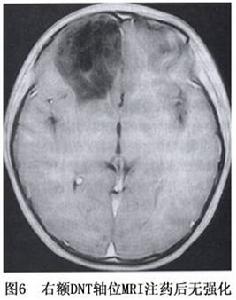 圖6
圖6無特殊表現。
其它輔助檢查:
神經節膠質瘤在CT上表現呈多樣性,大多數為低密度或等密度,少數為高密度,腫瘤邊界清鈣化或囊變各約1/3半數注藥後可出現混雜性增強,腫瘤對腦組織占位效應不明顯水腫少見,但位於大腦皮質表面的腫瘤可使顱骨內板受壓而局部變薄。MRI檢查T1加權像為低信號、T2為高信號、邊界清晰的占位影,病灶周圍腦回可有腫脹(圖1)。鈣化發生率不到1/10。約10%在頭顱平片中可發現鈣化影。
小腦發育不良性神經節細胞瘤CT為低密度或低與等密度相交替的混雜密度可累及一側小腦半球,偶有鈣化,有輕度占位效應,可幕上腦室擴大及四腦室輕度移位。MRI可見小腦半球異常增大,腫瘤無明顯占位效應,T1像為沿小腦溝排列的低信號和等信號的分層結構T2像為高信號和等信號交錯形成“虎紋征”(圖2,3)。注藥無強化。
治療
神經節膠質瘤手術切除為治療的主要措施。雖然有囊變,腫瘤仍以實質性為主,瘤內血供一般常有鈣化團大部分可做到腫瘤全切但部分腫瘤雖然表面邊界清,其深部界限常不確切,盲目追求全切易損傷深部結構,因此只可行次全切除。腫瘤對放療及化療均不敏感,即使腫瘤次全切除,亦不做常規放療等輔助治療。
小腦發育不良性神經節細胞瘤為良性病變,有局部占位效應,應手術切除,手術全切腫瘤可達到治療目的如切除不徹底則術後放療對防止復發可能有一定幫助。文獻報導有1例6年前曾手術,切除後未行放療,復發證實為本病。3例中術後放療者2例,因時間短(42個月及6個月)未見復發,也不好評價放療的作用;1例術後23個月未放療也未見復發。
對於胚胎髮育不良性神經上皮腫瘤手術切除來消滅致病灶是有效方法,手術目的是切除病灶控制癲癇發作可作病灶全切除,或是對發育不良的皮質及部分病灶切除,一般術後癲癇能完全消失,不需術後放療或化療,
預後
神經節膠質瘤預後好,5年生存率在90%以上,但應每年做一次CT或MRI檢查如有復發可再次手術。
胚胎髮育不良性神經上皮腫瘤預後好,很少復發,不影響病人生存。
