
引物
 引物
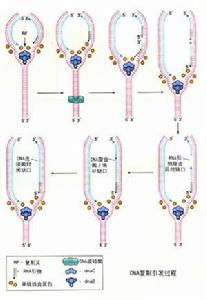
引物引物 (primer),台灣稱作引子,是一小段單鏈DNA或RNA,作為DNA複製的起始點,存在於自然中生物的DNA複製(RNA引物)和聚合酶鏈式反應(PCR)中人工合成的引物(通常為DNA引物)。
類型
存在有自然中生物的DNA複製引物(RNA引物)和聚合酶鏈式反應(PCR)中人工合成的引物(通常為DNA引物)。一般所說引物,指DNA引物,以下簡稱引物。
引物設計原則
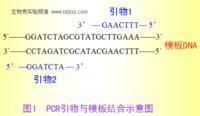
引物是人工合成的兩段寡核苷酸序列,一個引物與感興趣區域一端的一條DNA模板鏈互補,另一個引物與感興趣區域另一端的另一條DNA模板鏈互補。
在PCR(聚合酶鏈式反應)技術中,已知一段目的基因的核苷酸序列,根據這一序列合成引物,利用PCR擴增技術,目的基因DNA受熱變性後解鏈為單鏈,引物與單鏈相應互補序列結合,然後在DNA聚合酶作用下進行延伸,如此重複循環,延伸後得到的產物同樣可以和引物結合。
PCR引物設計的目的是找到一對合適的核苷酸片段,使其能有效地擴增模板DNA序列。如前述,引物的優劣直接關係到PCR的特異性與成功與否。對引物的設計不可能有一種包羅萬象的規則確保PCR的成功,但遵循某些原則,則有助於引物的設計。
1.引物最好在模板cDNA的保守區內設計。DNA序列的保守區是通過物種間相似序列的比較確定的。在NCBI上搜尋不同物種的同一基因,通過序列分析軟體(比如DNAman)比對(Alignment),各基因相同的序列就是該基因的保守區。
2.引物長度一般在15~30鹼基之間。
引物長度(primerlength)常用的是18-27bp,但不應大於38,因為過長會導致其延伸溫度大於74℃,不適於TaqDNA聚合酶進行反應。
3.引物GC含量在40%~60%間,Tm值最好近72℃
GC含量(composition)過高或過低都不利於引發反應。上下游引物的GC含量不能相差太大。另外,上下游引物的Tm值(meltingtemperature)是寡核苷酸的解鏈溫度,即在一定鹽濃度條件下,50%寡核苷酸雙鏈解鏈的溫度。有效啟動溫度,一般高於Tm值5~10℃。若按公式Tm=4(G+C)+2(A+T)估計引物的Tm值,則有效引物的Tm為55~80℃,其Tm值最好接近72℃以使復性條件最佳。
4.引物3′端要避開密碼子的第3位。
如擴增編碼區域,引物3′端不要終止於密碼子的第3位,因密碼子的第3位易發生簡併,會影響擴增的特異性與效率。
 引物
引物5.引物3′端不能選擇A,最好選擇T。
引物3′端錯配時,不同鹼基引發效率存在著很大的差異,當末位的鹼基為A時,即使在錯配的情況下,也能有引發鏈的合成,而當末位鏈為T時,錯配的引發效率大大降低,G、C錯配的引發效率介於A、T之間,所以3′端最好選擇T。
6.鹼基要隨機分布。
引物序列在模板內應當沒有相似性較高,尤其是3’端相似性較高的序列,否則容易導致錯誤引發(Falsepriming)。降低引物與模板相似性的一種方法是,引物中四種鹼基的分布最好是隨機的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不應超過3個連續的G或C,因這樣會使引物在GC富集序列區錯誤引發。
7.引物自身及引物之間不應存在互補序列。
引物自身不應存在互補序列,否則引物自身會摺疊成髮夾結構(Hairpin)使引物本身復性。這種二級結構會因空間位阻而影響引物與模板的復性結合。引物自身不能有連續4個鹼基的互補。
兩引物之間也不應具有互補性,尤其應避免3′端的互補重疊以防止引物二聚體(Dimer與Crossdimer)的形成。引物之間不能有連續4個鹼基的互補。引物二聚體及髮夾結構如果不可避免的話,應儘量使其△G值不要過高(應小於4.5kcal/mol)。否則易導致產生引物二聚體帶,並且降低引物有效濃度而使PCR反應不能正常進行。
8.5′端和中間△G值應相對較高3′端較低
△G值是指DNA雙鏈形成所需的自由能,它反映了雙鏈結構內部鹼基對的相對穩定性,△G值越大,則雙鏈越穩定。應當選用5′端和中間△G值相對較高,而3′端△G值較低(絕對值不超過9)的引物。引物3′端的△G值過高,容易在錯配位點形成雙鏈結構並引發DNA聚合反應。(不同位置的△G值可以用Oligo6軟體進行分析)
9.引物的5′端可以修飾,而3′端不可修飾。
引物的5′端決定著PCR產物的長度,它對擴增特異性影響不大。因此,可以被修飾而不影響擴增的特異性。引物5′端修飾包括:加酶切位點;標記生物素、螢光、地高辛、Eu3+等;引入蛋白質結合DNA序列;引入點突變、插入突變、缺失突變序列;引入啟動子序列等。
引物的延伸是從3′端開始的,不能進行任何修飾。3′端也不能有形成任何二級結構可能。
10.擴增產物的單鏈不能形成二級結構。
某些引物無效的主要原因是擴增產物單鏈二級結構的影響,選擇擴增片段時最好避開二級結構區域。用有關軟體(比如RNAstructure)可以預測估計mRNA的穩定二級結構,有助於選擇模板。實驗表明,待擴區域自由能(△G°)小於58.6lkJ/mol時,擴增往往不能成功。若不能避開這一區域時,用7-deaza-2′-脫氧GTP取代dGTP對擴增的成功是有幫助的。
11.引物應具有特異性。
引物設計完成以後,應對其進行BLAST檢測。如果與其它基因不具有互補性,就可以進行下一步的實驗了。
值得一提的是,各種模板的引物設計難度不一。有的模板本身條件比較困難,例如GC含量偏高或偏低,導致找不到各種指標都十分合適的引物;用作克隆目的的PCR,因為產物序列相對固定,引物設計的選擇自由度較低。在這種情況只能退而求其次,儘量去滿足條件。
做RealTime時,用於SYBRGreenI法時的一對引物與一般PCR的引物,在引物設計上所要求的參數是不同的。引物設計的要求:
●避免重複鹼基,尤其是G.
 引物
引物●Tm=58-60度。
●GC=30-80%.
●3'端最後5個鹼基內不能有多於2個的G或C.
●正向引物與探針離得越近越好,但不能重疊。
●PCR擴增產物長度:引物的產物大小不要太大,一般在80-250bp之間都可;80~150bp最為合適(可以延長至300bp)。
●引物的退火溫度要高,一般要在60度以上;要特別注意避免引物二聚體和非特異性擴增的存在。
而且引物設計時應該考慮到引物要有不受基因組DNA污染影響的能力,即引物應該跨外顯子,最好是引物能跨外顯子的接頭區,這樣可以更有效的不受基因組DNA污染的影響。做染料法最關鍵的就是尋找到合適的引物和做污染的預防工作。對於引物,你要有從一大堆引物中挑出一兩個能用的引物的思想準備---尋找合適的引物非常不容易。
關於BLAST的作用應該是通過比對,發現你所設計的這個引物,在已經發現並在GENEBANK中公開的不物種基因序列當中,除了和你的目標基因之外,還有沒有和其他物種或其他序列當中存在相同的序列,如和你的目標序列之外的序列相同的序列,則可能擴出其他序列的產物,那么這個引物的特異性就很差,從而不能用。
常用引物設計軟體
■Oligo 6
Oligo6是目前使用最為廣泛的一款引物設計軟體,除了可以簡單快捷地完成各種引物和探針的設計與分析外,還具有很多其他同類軟體所不具有的高級功能:
●已知一個PCR引物的序列,搜尋和設計另一個引物的序列;
●按照不同的物種對MM子的偏好性設計簡併引物;
 引物設計軟體
引物設計軟體●對環型DNA片段,設計反向PCR引物;
●設計多重PCR引物;
●為LCR反應設計探針,以檢測某個突變是否出現;
●分析和評價用其他途徑設計的引物是否合理;
●同源序列查找,並根據同源區設計引物;
●增強了的引物/探針搜尋手段。設計引物過程中,可以"Lock"每個參數,如Tm值範圍和引物3’端的穩定性等;
●以多種形式存儲結果;支持多用戶,每個用戶可保存自己的特殊設定。
■ Premier 5.0
PrimerPremier5.種用來幫助研究人員設計最適合引物的套用軟體利用它的高級引物搜尋引物資料庫巢式引物設計引物編輯和分析等功能可以設計出有高效擴增能力的理想引物也可以設計出用於擴增長達50kb以上的PCR產物的引物序列。
該軟體主要由GeneTank序列編輯,Primer引物設計,Align序列比較,Enzyme酶切分析和Motif基序分析等幾個主要功能板塊組成。
