
基本內容
RAPD(random amplified polymorphic DNA)即隨機擴增多態性DNA標記,其基本原理與PCR技術一致。
DNA製備
 RAPD分析軟體
RAPD分析軟體參照薩姆布魯克等(1996) 的方法略加改進,取足肌約100 mg 切碎並溶於裂解液中,加入蛋白酶K 至終濃
度為100μg/ ml,充分混勻後37 ℃消化4~5 h。加入RNase 至終濃度100μg/ ml,充分混勻後60 ℃作用30min。向樣品中依次加入等體積的飽和酚、酚/ 氯仿/ 異戊醇(25 :24 :1) 和氯仿/ 異戊醇(24 :1) 抽提蛋白,然後以兩倍體積的冰冷無水乙醇沉澱,DNA 乾燥後加入適量TE 溶解,4 ℃保存。在UNIC22100 型紫外分光光度計上測定DNA 純度及估計模板濃度,並用瓊脂糖凝膠電泳檢測DNA 分子量。
擴增
使用PE29600 型PCR 擴增儀,反應總體積為25μl,其中10 ×Buffer,2 mmol/ L Mg,0.12 mmol/ L 引物,0.12 mmol/ L dNTPs,2U Taq 酶,20 ng 模板DNA。
PCR 反應程式為95 ℃預變性5 min,94 ℃變性45 s,36 ℃復性45 s,72 ℃延伸90 s,30 個循環後,72 ℃延伸10 min,4 ℃保存。
每次PCR 反應均設不含模板DNA 的空白對照。
擴增產物以1.2 %瓊脂糖凝膠電泳分離,溴化乙錠染色,在BioRad2 Gel2700 TM 凝膠成像系統下觀察並拍照記錄。
數據處理
將RAPD 電泳譜帶位點上有擴增位點的記為1,無擴增位點的記為0,建立原始數據矩陣,用Pop Gene
(Ver1132) 軟體進行數據統計。群體的多態位點百分率P = 擴增的多態位點數/ 擴增的總位點數×100 %。
群體遺傳多態度用Shannon′s 多樣性指數(H0) 表示,按照Wachira 等(1995) 的公式,Shannon′s 多樣性指數
H0 = - ΣXi ln (Xi / n),式中,X i 為位點i 在某一群體中出現的頻率,n 為該群體檢測到的位點總數; H0 可以分
解為群體內遺傳多態度Hpop 和群體遺傳多態度總量Hsp,其中,Hpop = ΣH0 / n(n 為所檢測的群體數) ; Hsp = -
ΣX (X 為n 個群體的綜合表型頻率)。
概述
運用隨機引物擴增尋找多態性DNA片段可作為分子標記。這種方法即為RAPD(RandomamplifiedpolymorphicDNA,隨機擴增的多態性DNA)。儘管RAPD技術誕生的時間很短,但由於其獨特的檢測DNA多態性的方式以及快速、簡便的特點,使這個技術已滲透於基因組研究的各個方面。該RAPD技術建立於PCR技術基礎上,它是利用一系列(通常數百個)不同的隨機排列鹼基順序的寡聚核苷酸單鏈(通常為10聚體)為引物,對所研究基因組DNA進行PCR擴增.聚丙烯醯胺或瓊脂糖電泳分離,經EB染色或放射性自顯影來檢測擴增產物DNA片段的多態性,這些擴增產物DNA片段的多態性反映了基因組相應區域的DNA多態性。
RAPD所用的一系列引物DNA序列各不相同,但對於任一特異的引物,它同基因組DNA序列有其特異的結合位點.這些特異的結合位點在基因組某些區域內的分布如符合PCR擴增反應的條件,即引物在模板的兩條鏈上有互補位置,且引物3'端相距在一定的長度範圍之內,就可擴增出DNA片段.因此如果基因組在這些區域發生DNA片段插入、缺失或鹼基突變就可能導致這些特定結合位點分布發生相應的變化,而使PCR產物增加、缺少或發生分子量的改變。通過對PCR產物檢測即可檢出基因組DNA的多態性。分析時可用的引物數很大,雖然對每一個引物而言其檢測基因組DNA多態性的區域是有限的,但是利用一系列引物則可以使檢測區域幾乎復蓋整個基因組。因此RAPD可以對整個基因組DNA進行多態性檢測。另外,RAPD片段克隆後可作為RFLP的分子標記進行作圖分析。
試驗條件
1. 藥品試劑
模板DNA(10~100ng)
寡核苷酸引物(20mmol/L貯存液,可向Operon Technologies或Perkin Elmer公司購買,也可以自己合成)
 RAPD
RAPD0.2 mmol/L dNTPs(必須使用高質量的dNTPs,這一點非常重要。dNTPs經反覆化凍後會發生降解,因此應分成小份保存。應注意混合物中四種dNTP的量要相等)
Taq DNA聚合酶(Perkin Elmer, Norwalk, Connecticut)
10×PCR緩衝液(500 mmol/L KCl, 15 mmol/L MgCl2, 100 mmol/L Tris•HCl, pH 8.3)
礦物油
電泳所需試劑
2. 儀器設備
PCR擴增儀
電泳裝置
微量離心機
微量移液器(1~20ml和20~200ml)
0.5 ml Eppendorf管
紫外線觀察裝置及照相設備
操作程式
(1)在冰里向無菌的Eppendorf管中加入以下反應物:
| 水 | 14.75 ml |
| 10×緩衝液 | 2 ml |
| 10×dNTPs | 2 ml |
| 引物(20 mmol/L) | 0.2 ml |
| Taq DNA聚合酶 | |
| 終體積 | |
| DNA(10~100ng) | 1 ml |
| 石蠟油 | 20ml |
(2)93℃反應2 min後開始如下循環:
93℃變性反應1min
36℃退火反應1min
72℃延伸反應1.5min
經過45個循環後,最後一個循環72℃增加5min,循環結束後反應產物置於4℃保存。
(3)20ml反應產物走凝膠電泳,經溴化乙錠染色檢測擴增的情況。
解決方法
擴增偏差或無擴增
――在部分或全部管中缺少一個組分。重複少量幾個反應以確定所有的PCR組分是否都已加入。
 RAPD擴增圖
RAPD擴增圖――PCR抑制物可能與DNA一起純化,改變DNA的濃度。
――在DNA分離中加入一個清洗步驟(如苯酚抽提)。
――稀釋新的DNA溶液。
――引物母液失效。重新配製母液,使用不同的引物或提高引物濃度。
――延長退火時間(在PCR程式中的第二部分),或降低升溫轉換步驟之間的速率。
――提高每個反應中的Taq聚合酶的濃度。
――補加新的組分,該滅菌的都高壓滅菌。
結果難以辨認
――更換Taq聚合酶緩衝液。
――檢查引物(使用另外一個引物,或者將引物末端標記,在16% 6mol/L尿素聚丙烯醯胺膠上電泳檢測)。
――檢查Taq聚合酶活性(與不同批量的酶作比較)。
――改變DNA濃度。
質量產物彌散狀分布
――降低DNA濃度。
――降低Taq聚合酶濃度。
――減少凝膠上樣量。
――可能是由於循環數過多的結果(Bell和Demarini 1991),減少循環數。
――確認是否使用正確的緩衝液(不是水!)製備凝膠。
――在較低的電壓下電泳。
單一帶
在對照和所有檢測樣品中均為一條很強的單一帶
――確認引物序列不是迴文結構。這可能是引物多聚體造成的結果。使用不同的引物。
帶譜不可重複
――DNA過多或過少。把基因組DNA濃度控制在10~100ng範圍之內,DNA量太少時,“真正”靶序列與引物的結合效率低,因此引物擴增會產生假的條帶,這就稱為引物偽跡;DNA量太多會導致錯誤配對(指引物與基因組DNA的配對)。每個樣品進行兩個不同DNA濃度的反應。
――只考慮主要條帶。
染色後凝膠背景太強
――減少染色時間。
――凝膠脫色時間延長。
――PCR反應中DNA含量或Taq聚合酶過多。
質量產物分離不充分
――在更高濃度的瓊脂糖凝膠、專業純凝膠或聚丙烯醯胺凝膠上分離產物。
技術問題及對策
RAPD技術是由多種成分參加的生化反應,反應中各種成分均為微量,儘管其反應靈敏度高,但是影響因素較多,會出現重複性差等一些問題。為了得到較穩定的結果,各種反應參數必須事先最佳化選擇,操作中每一步都必須小心謹慎,以防止出現差錯。
1 溫度
RAPD一般反應條件是:變性92-95℃,常用94℃,30-60s;延伸72℃,60-120s;退火35-37℃,30-60s。變性和延伸溫度一般沒有太大變化,而退火溫度影響較大。退火溫度低,引物和模板結合特異性較差,出現的條帶可能增多;退火溫度高,引物和模板結合特異性增加。有人提出,在尋找基因差異為目的的實驗中,退火採用33-34℃效果更好。在最佳化方案中,退火溫度可能要提高,以便降低背景,得到少而清晰的"帶"。溫度的梯度率也是十分重要的,特別是退火與延伸之間的梯度尤為重要。各種儀器的控溫、升降溫性能均有差別,一些儀器(如一些舊型號的儀器)在到達特定的溫度前就開始計時,一些PCR儀顯示的溫度與實際溫度會有差異。這些在設計程式和結果分析時有必要考慮,所以註明所用儀器的生產廠家、品牌、規格,就顯得很有必要,對於同一批實驗,注意控制在同樣的溫度條件下進行反應,結果才有可比性。
2 反應物的影響
PCR擴增受多種成分的制約,產物僅在部分循環中呈指數式(2)增長,逐漸將達到一個平台期,模板是關鍵制約因素之一,RAPD產物取決於此,實驗中非常精確的模板濃度是十分關鍵的一個步驟。濃度過低,分子碰撞機率低,偶然性大,擴增產物不穩定;濃度過高,會增加非專一性擴增產物。更重要的是,若循環數控制不好,引物過早消耗完,後面循環中,PCR擴增產物的3端會和原模板及每一次循環的擴增產物退火,造成不等長度的延伸。因此,如果原模板濃度過高,非專一性擴增產物就足以形成背景,這是高濃度模板造成彌散型產物的原因。相對而言,模板純度影響不大,即反應液中蛋白質、多糖、RNA等大分子物質影響不大。理想的模板和引物濃度能產生多態性豐富、強弱帶分明的RAPD圖譜。引物3端的重要性似乎更大。此外,Mg+2濃度也影響反應的參數包括產物的特異性和引物二聚體的形成,人們在各自的實驗中得出不同的結論,大約在1.5-4mmol/L範圍內。總之,各種反應物的濃度應事先進行篩選最佳化。
3 污染問題
實驗中應設空白對照,因為引物、各種緩衝液和雙蒸水都會造成污染。而且Taq酶也可能出現污染,給分析帶來困難,所以必須設定對照以排除系統誤差。並且,酶也儘可能最佳化。
4 擴增條帶的取捨
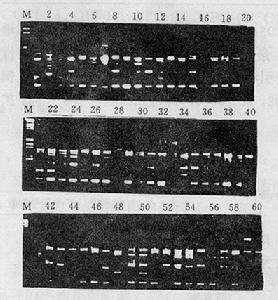 RAPD
RAPD重複性問題 為了克服RAPD重複性差的問題,應設定重複實驗。作為DNA多態性標記位點的條帶是應該可重複的,重複性好是最重要的取捨指標,而帶的強弱不應該作為取捨的指標。有學者認為帶的強弱與其在基因組中的拷貝數有關。但據Thormann對十字花科植物分析,RAPD產物的強弱與該產物在基因組中的拷貝數無直接關係,而與引物及模板的同源性程度有關。分析一個位點時,要作全面分析,若某一個體的全部帶均弱,其模板可能有問題(濃度、分子量);若某一個體的大部分帶與其它個體強度一致,僅有一條或少數幾條帶弱,可能存在拷貝數差異。重複性不好的弱帶(無規律出現的帶)可能由非專一性擴增、產物間退火或其它人為因素造成,不能記錄。
異源雙鏈問題 Ayliffe研究亞麻鏽病菌時發現F1代中出現的一條帶在親本中沒有出現,進一步研究發現這一條帶是由2個等位基因組成的異源雙鏈形成的,這2個等位基因除中間插入的38bp的序列外,其餘序列相同。這種條帶可以在後代中遺傳,在該研究中這種條帶大約占0.16%。在蜜蜂的研究中也發現了相似的問題,這種條帶可以用單鏈核酸酶將其消除。也可以將電泳溫度提高到60℃以上,使異源雙鏈變性,以消除其影響。
非孟德爾遺傳的條帶 Heun等分析認為,在顯示多態性的非模糊條帶中有92.5%的表現為孟德爾顯性遺傳,其餘的條帶不符合孟德爾遺傳,分析認為可能由不同的序列組成。一般實驗中大多採用符合孟德爾遺傳的條帶進行分析。在連鎖圖譜分析中,基因型錯誤可以部分消除,即在F1代或1:1混合物中沒有出現的條帶,在其父母本中應儘可能去掉。
同一條帶的同源性問題 Thormann將RAPD產物標記作探針,雜交結果表明,與探針片斷遷移率一致的帶有時並不同源,這種情況僅發生在種間。並比較了RFLP和RAPD標記在種內和種間得出的結果,發現RFLP和RAPD標記在種內水平的結果完全吻合,但在種以上等級的結果相差甚遠,這說明在電泳結果的同一個帶中,有可能存在序列不同但分子量相同的幾條帶,RAPD無法檢測。這種可能在種內較少發生,而這種間可能性較大,Castagna用RFLP和RAPD比較研究也得出了相同的結論。
5 電泳解析度問題
電泳時,基因組不同位置的擴增產物共同移動的可能性是有的,即不同分子量的擴增產物可能在電泳時沒有分開而形成一條帶,凝膠分離系統和基因組的親緣關係有可能提高這種機率。一般而言,聚丙烯醯胺和銀染的分辨效果比瓊脂糖和溴化乙銨要高,用較長(可達到20cm)或濃度更高(2%)的瓊脂糖凝膠有助於提高解析度。當然,隨著解析度和靈敏度的提高,更可能出現實驗誤差。所以有人評述:最保險而簡單的方法是用瓊脂糖電脈分離而且只注意那些無疑而重發性高的帶。但是,聚丙烯醯胺和銀染所揭示的高信息量的多態性無疑可加快分子標記的鑑定過程。
6 顯性標記問題
RAPD標記來自於模板DNA的複製,經過30一40次循環,擴增條帶數量理論上可達2。原模板中擴增位點是純合還是雜合已無法判斷,因為純合位點的擴增條帶只相當於雜合位點的2倍,電泳時無法檢測到其差別。雜交中。如果親本一方為純合體,F1代就可以全部表現出該條帶;如為雜合體,該條帶在F1中應為l:l分離,即一半後代有該條帶,而另一半則沒有。當然來自於多拷貝區的條帶分析更為複雜。
套用問題
1 遺傳圖譜構建的問題
RAPD是一種顯性標記,符合孟德爾遺傳定律,不能區分雜合型和純合型,因此在遺傳分析及遺傳圖譜的構建等一些方面受到限制。目前,在實踐中,主要利用了以下2種方法。
①利用單倍體 利用胚乳進行RAPD分析,可以克服構建遺傳圖譜中這些困難。因為針葉樹的胚乳是單倍體,記錄了雜合的母本染色體組在減數分裂過程中的遺傳事件,不存在雜合型,進行RAPD遺傳分析時,與共顯性標記具有同樣的遺傳行為,是天然的優良作圖材料。現在已進行遺傳圖譜構建的有濕地松、歐洲赤松、火炬松、楊樹、日本柳杉和桉樹。但是由於缺乏細胞遺傳學的研究,暫時還不能確定連鎖群和染色體之間的對應關係。
②將RAPD條帶作為單劑量標記,單劑量標記(Single-dose RAPD markers)是從兩物種和其雜交產生F1代的分子標記中選擇的。選擇構建圖譜用的標記有2個原則:A、它們必須在一個親本中存在而在另一個親本中不存在;B、它們在後代中必須以l:1分離。這種方法相當於一個雜合體和一個隱性純合體的測交,稱為雙向假測交(Two-way pseudo-testcross)。在多倍體植物中,不論基因組是如何組成的(異源多倍體或同源多倍體),也不論材料的多倍性水平如何,單劑量的標記都相當於簡單的等位基因(同源多倍體)或相當於二倍體基因座上的雜合等位基因(異源多倍體)。因此根據這些單劑量標記的連鎖情況可以構建分子圖譜。RAPD條帶作為單劑量標記使那些基因組DNA含最大,世代周期長的喬木和多倍體植物的遺傳圖譜研究走出了幾乎停滯的狀態。Grattapagalia利用這種策略首先構建了巨桉和尾葉桉的分子連鎖圖譜。Conner利用這種策略構建了3個蘋果品種的分子圖譜。Mudge利用甘蔗及雜交後代的RAPD條帶作為單劑量標記進行連鎖分析構建了甜根子草的51個連鎖群,並分析了其染色體組的數目。雖然這些連鎖圖譜是個體特異性的,但可以為數量性狀定位提供框架結構,為標記輔助選擇育種奠定基礎。
2 系統學研究中的問題
 RAPD
RAPD系統學是RAPD研究最為迅速的領域,許多作物都用RAPD作過親緣關係分析,大部分結果和形態分類結果相類似,僅有少數例外。但是近年來,有人對其結果的可靠性提出不同的看法,主要有以下幾點: ①由於引物的競爭性等,基因組的RAPD位點有時不能全部檢出,造成假象上的差異。根據這種情況,可以認為,RAPD可以套用於種間乃至近緣屬之間關係的研究,但有一定的局限性。因不同種度的擴增產物難以作同源性分析,只能作表征分析,從而只能從相似性或遺傳距離上去分析親緣關係。其結果可能與真實的系統關係相左。
②因RAPD在種間或屬間的變異水平很高,取樣代表性是一個較大的問題,即利用某一個體代表一個種或用一個種代表一個屬都可能造成結果的偏差。因而,最好能找到群體特異或種特異性的條帶。
③在表征分析中另一個值得注意的問題是一些RAPD產物的出現存在相關關係,即一個位點與另一個位點相連鎖。若將這個差異單獨計算,相當於對某一性狀極度加權。
④在系統發育學分析中,因為RAPD條帶具有顯性的遺傳特徵,多態性信息量較低。如果其符合二歧分枝樹的假定,就可以對變化的性狀予以加權,並進行分析。但是許多植物的核基因組是有性的二倍體,而且它們的進化歷史是網狀分枝樹,不便於分析。通常認為葉綠體、線粒體和有些細菌及真菌的病原體符合二歧分枝樹的假定。但由於其基因組較小,沒有重組,它們的信息量只相當於核基因組中具有多個等位基因的一個同工酶座位,這樣估算的基因多樣性值的標準差理論上會比有多個核基因蛋白座位得出的大。其非編碼區在一些動物類群中的進化速度很快,因此做種上水平研究時將會影響結果的準確性,但種下水平影響較小。
⑤RAPD和農藝性狀的系統分析比較說明,二者相差不大,沒有出現相反的情況。而且RAPD比農藝性狀提供了更多的遺傳信息。
總之,用RAPD作系統分析應結合其它方法,結果才更有說服力。
3 標記、定位目的基因
標記、定位目的基因是相對較為緩慢的一個領域,這是因為RAPD標記探測的是整個基因組的變化,包括編碼區和非編碼區,要和基因位點聯繫起來有一定的困難。一般的變異群體性狀的變化涉及到基因組內較大的範圍(微效多基因),RAPD不可能全部檢出。而且RAPD檢出的多態性又不一定是性狀變異的標誌(可能擴增於非編碼區)。另一困難是RAPD擴增的特異性片段有些為重複序列,轉化為RFLP探針有一定困難。據Grattapaglia用48個RAPD片段所作的雜交實驗表明,有53%來自於低拷貝區(1000)。這隻有通過篩選出與單拷貝區連鎖的標記予以解決。如果RAPD標記和基因位點或性狀不能聯繫起來,其實用價值就會大大降低。為此人們作了以下探索。
①利用易位系、回復突變系、等位基因系、近交重組系等特殊材料 從理論上講,除目的基因及鄰近區域外,其它部分完全一致。如果檢測出多態性,差異必定在目的基因及鄰近區中,也就是說RAPD標記在這個範圍內與目的基因連鎖。但是要獲得這些材料比較困難,例如等位基因系需要回交20代,時間太長。RAPD分析中一般用近等位基因系NILs(Near isogenic lines),回交6代以上即可。回交6代時,供體DNA的比例為l/128,用RAPD找到的與目的基因連鎖的標記與目的基因在距離上還相當遠。
②集團分離法(BSA)對不存在上述特殊生物模型的群體,可採用這種方法。即利用F2分離群體為材料,根據目的基因的表型把F2群體分為2群(例如抗病的和不抗的),每一群中將各個體DNA等量混合,這樣形成2個DNA混合池。在每個基因池中,不管群體性狀如何,在目的基因的表型上都是一致的,2個群體之間,目的基因的表型都是相反的。因此,在2個群體之間完全不同、在每個群體之內各個體間又是完全相同的RAPD擴增產物即可認為是與目的基因連鎖的RAPD標記。這種方法相當於把分析材料作為近等位基因系。
在找到合適的分子標記後,既可直接用於輔助選擇育種(Molecule-marker assisted selection,MAS),也可通過對特異片段的分離和兩端測序將之轉換為序列標誌位點(Sequence-tagged sites,STS)。從這個分子標記出發找到目的基因需經過幾個步驟:A、用稀有切點內切酶將基因組DNA切成大片斷DNA;B、用脈衝電泳分離大片斷DNA;C、將大片斷DNA克隆到YAC(Yeast artificial chromosome)中,建立YAC文庫;D、以顯示多態性的擴增產物為探針從YAC文庫中調出含有該RAPD標記和與其連鎖的目的基因的大片斷DNA;E、以該RAPD標記為起點向目的基因進行染色體滑步或跳躍,以找到目的基因。Michelmore首先利用這種方法找到了萵苣抗性基因的標記,Koller等利用這種方法找到與蘋果瘡痂病抗性基因有關的分子標記。
③利用一套缺體一四體系或雙端體系,以獲得與目的基因連鎖的標記引物進行擴增反應,然後進行原位雜交,確定特異性帶在染色體上的位置。
這些工作在一些基礎研究較多的領域或與人類關係較為密切的物種中進展較為迅速,比如小麥、水稻找到的RAPD分子標記已有許多。但林木等多年生植物研究進展稍遲滯,關鍵是缺乏合適的群體。
4 純度和外源基因檢驗問題
雖然由於RAPD條帶的靈敏和重複性等問題,純度檢驗研究還只停留在實驗階段。但已有的對大麥、玉米、大豆、棉花、小麥、水稻的實驗表明,RAPD技術是鑑定品種的有效方法。相信其在純度檢驗和專利保護方面會取得較為廣泛的套用。
藥材鑑定
植物藥材相近種類的鑑定
 轉基因草類
轉基因草類邵鵬柱等在1994 年首次報導了對中藥材人參與西洋參採用Ap2PCR 標記進行鑑別,他們採用20 個鹼基的Galk ,Seq2 ,24 個鹼基的MB forward ,MB reverse 和27 個鹼基的OTCSbackward 作引物成功地利用AP2PCR 指紋圖譜技術鑑定出人參和西洋參。1995 年分離人參屬三種植物人參、西洋參、三七(P. notoginseng) 和四種偽品包括桔梗、紫茉莉、櫨蘭、商陸的基因組DNA ,分別採用10 個鹼基的OPC25 和OPC220 作引物,用RAPD 標記可以明顯地區別,說明DNA 分子也能區別人參屬三個品種和人參及其偽品。中國南方部分省區有多種菊科不同屬的植物如地膽草、一點紅、苦蕒菜、毛大子草、山萵苣、苦苣菜以“土公英”混作蒲公英使用。曹暉等利用Ap2PCR 和RAPD分子標記技術對蒲公英及六種土公英混淆品進行了DNA 指紋鑑別研究,結果顯示它們的DNA 指紋圖譜存在明顯差異,利用這種差異可以從分子水平上
鑑別蒲公英及其混淆品。Kohjyouma M 等採用CTAB 法提取菊科蒼朮屬五種植物茅蒼朮、北蒼朮、單葉蒼朮、關蒼朮和白朮等新鮮葉片組織的基因組DNA ,利用RAPD 技術獲得了清晰的多態性DNA 指紋圖譜,能明顯地區分蒼朮的幾個品種,發現在基因水平上單葉蒼朮與茅蒼朮十分接近,而與白朮和關蒼朮相距較遠,後兩者與北蒼朮又相近似。這一結果與RFL P (Restriction fragmentlength polymorphism) 方法得到的結果相近。說明RAPD 能有效地鑑別蒼朮屬植物。
植物藥材基原的鑑定
Yamazaki M 等採用酸性緩衝液提取法分離了4 種菊科甘草屬植物光果甘草、甘草、刺毛甘草和刺果甘草的基因組DNA ,通過對RAPD 指紋圖譜分析,表明光果甘草與甘草遺傳關係非常接近,刺毛甘草和刺果甘草的遺傳關係相距較遠,這種結果與RFL P 所得結果類似,也與植物分類學研究吻合。他們進一步對4 種商品甘草藥材(新疆甘草、西北甘草、西伯利亞甘草、阿富汗甘草) 採用RAPD 方法作遺傳距離估算,來對其進行基原鑑別,逐步證實了阿富汗甘草來源於光果甘草、西伯利亞甘草來源於甘草、新疆甘草和西北甘草來源於刺果甘草。曹暉等以CTAB 微量提取法分離菊科植物地膽草、白花地膽草和假地膽草以及四種商品苦地膽藥材的基因組DNA ,採用長引MBforward(24 個鹼基) ,GalK(20 個鹼基) 進行AP2PCR 擴增和用OPC26 (10 個鹼基) 的短引物進行RAPD 擴增,獲得清晰可靠的DNA 指紋圖譜,根據DNA帶型差異鑑別出地膽草及其混淆品。同時通過對DNA 指紋圖譜的相似度指數值計算, 證實四種商品藥材苦地膽的基原為菊科植物地膽草。
複方製劑、粉劑中藥材品種的鑑定
 RAPD分析樣品
RAPD分析樣品中藥材複方製劑、粉劑等由於其中的各種生藥外觀性狀完全遭到破壞,要了解某味藥是否存在,用傳統的鑑定方法有時難以奏效。但是使用RAPD 技術時,從理論上來講,如果只要能製備出該種藥材的高度特異
性的寡核酸引物,即可達到鑑定的目的。Ozeki 等採用GTC(異硫氰酸胍) 微量提取法分離人參屬三種植物人參、西洋參和竹節參以及兩種人參製劑高麗紅參泡茶OTANECHAN(日本的一種人參組織培養物製成的茶) 的基因組DNA ,通過RAPD 分析,結果發現人參的根毛組織和乾燥藥材產生相同的RAPD 指紋圖形,人參、西洋參和竹節參之間採用不同的引物產生不同的RAPD 指紋圖;高麗紅參泡茶中幾乎沒有DNA 片段擴增(其提取物無DNA 模板之故) , 但從OTANECHAN 中擴增到了與人參愈傷組織相同RAPD 指紋的DNA 片段。這表明了RAPD 技術套用於中藥傳統製劑中組分鑑別的可能性。黃璐琦等對來源於已於13 個種3 個變種的天花粉及其類似品共26 份樣品,用8 個擴增多態性好的引物分別進行擴增,得到清晰、穩定的條帶83 條,然後採取聚類分析的方法。結果表明全部樣品可被有效地分為三大類:第一類是大宗商品和小宗商品,第二類是天花粉藥材商品中極易混淆的湖北栝樓根和紅花栝樓根,第三類全部是混淆品和地區習慣用藥。對不同植物來源的天花粉,尤其是對來自不同組或組上水平的天花粉采RAPD 技術能夠很好地把它們區別開來,其結果與物種間的親緣關係基本一致。這為解決粉末及破碎藥材的鑑定提供了新的方法。
動物藥材的鑑定
這方面的文獻報導比較少。王義權等成功地將RAPD 技術套用於六種蛇類的基因組DNA的多態性檢測,在20 個引物中找出16 個引物產生了253 個擴增片段,準確地區別了游蛇科5 個種和蝰蛇科1 個種,片段分布和片段共享度聚類分析所得的游蛇科5 種蛇之間的親緣關係與染色體、顱骨和陰莖研究的結果基本一致。顯示DNA 分子標記技術可以成為一種準確鑑別蛇類藥材的新方法,RAPD 同樣適合於動物中藥材的鑑定。
藥材的野生類型與栽培品種的鑑定
由於生態環境的改變和人們生產活動的影響,中藥材遺傳特性及其生藥品質在一定程度上產生了變化,產地和栽培也對生藥品質和藥性產生巨大影響。在人類回歸自然呼聲日益高漲的今天,人們普遍認為野生品比栽培品要好。同時,由於經濟利益的驅使,以栽培品冒充野生品的現象時有發生,給臨床造成混亂。因野生品和栽培品來自同一個物種,植物形態、藥材性狀、化學成分、生物活性等往往無明顯差異,這就需要尋找一種有效的方法進行鑑別,RAPD 等分子標記的產生為這種鑑定提供了良好的套用前景。馬小軍等利用RAPD、A FL P 標記對人參、圓參與山參品種的研究中發現,RAPD、AFLP 標記可以用於野生與栽培品種的鑑別,其長脖類型更接近野生人參。
道地藥材鑑定
道地藥材是人們在長期的臨床實踐中,經臨床總結作為評價藥材質量的綜合標準。它是一種長期進化和生態適應過程中,某一物種的變種、生態型、品種或居群適應於特定的生態地理環境條件所形成的優良種質資源。道地藥材的研究,包括其形成與鑑定,過去主要集中於化學、藥理研究,從DNA 標記水平的報導很少。胡珊梅等用RAPD技術探討澤瀉道地藥材與非道地藥材之間遺傳變異的大小, 並建立了道地藥材的品質鑑定方法,為道地藥材研究提供了有益的啟示。可以預見,藉助分子標記,對道地藥材的快速、準確鑑定,無疑會給我們提供新思維、新視野。
