
主要分類
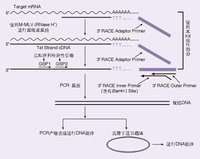
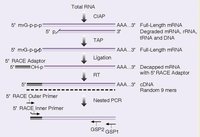 RACE技術
RACE技術一般分5-和3-RACE兩種。
3-RACE較簡單,首先將mRNA或總RNA用PolyT引物反轉錄,根據一般基因具有polyA尾巴的特點,選用特異引物(根據已知序列設計)和PolyT引物PCR即可。大多實驗者反映一次PCR可以搞定。
5-RACE相對較難,目前流行幾種5-RACE。其一為加接頭(傳統),根據接頭引物和自己設計特異引物PCR,可以設計巢式PCR二次擴增。另外,有利用反向PCR技術,連線成環在PCR。還有,GENE公司一種smartRACEPCR,利用反轉酶末斷加C特點,直接加上多G接頭,轉換模板而無需用連線酶加接頭。
發展歷史
 RACE技術
RACE技術經典的RACE技術是由Frohman等(1988)發明的一項技術,主要通過RT-PCR技術由已知部分cDNA序列來得到完整的cDNA5’和3’端,包括單邊PCR和錨定PCR。該技術提出以來經過不斷發展和完善,克服了早期技術步驟多、時間長、特異性差的缺點(Frohman等,1995:Schaefer,l995:Chen,1998:Bespalova等,1998:Matz等1999)。對傳統RACE技術的改進主要是引物設計及RT-PCR技術的改進:改進之一是利用鎖定引物((lockdockingprimer)合成第一鏈cDNA,即在oligo(dT)引物的3′端引入兩個簡併的核苷酸【5′-Oligo(dT)16_30MN-3′,M=A/G/C;N=A/G/C/T】,使引物定位在poly(A)尾的起始點,從而消除了在合成第一條cDNA鏈時oligo(dT)與poly(A)尾的任何部位的結合所帶來的影響;改進之二是在5‘端加尾時,採用poly(C),而不是poly(A);改進之三是採用RNaseH一莫洛尼氏鼠白血病毒(MMLV)反轉錄酶或選擇嗜熱DNA聚合酶可能在高溫h(60度-70度)有效地逆轉錄mRNA,從而消除了5‘端由於高CC含量導致的mRNA二級結構對逆轉錄的影響;改進之四是採用熱啟動PCR(hotstartPCR)技術和降落PCR(touchdownPCR)提高PCR反應的特異性。 隨著RACE技術日益完善,目前己有商業化RACE技術產品推出,如CLONTECH的MarathoTM技術和SMARTTMRACE技術術。邢桂春等(2001)先後使用上述兩種試劑盒進行RACE反應,結果發現使用MarathonTM所得到的片斷總是比採用SMARTsup]TMRACE試劑盒到所得到的片斷短。其原因在於MarathonTM技術反轉錄反應往往不能真正達到mRNA的5’末端。所以認為,進行RACE反應應當優選SMARTTMRACE試劑盒。
引物設計
基因特異性引物(GSPs)應該是:
1、23-28nt
2、50-70%GC
3、Tm值≥65度,Tm值≥70度可以獲得好的結果需要實驗者根據已有的基因序列設計5‘和3‘RACE反應的基因特異性引物(GSP1和GSP2).由於兩個引物的存在,PCR的產物是特異性的。
4、cDNA的合成起始於polyA+RNA。如果使用其它的基因組DNA或總RNA,背景會很高。
5、RACEPCR的效率還取決於總的mRNA中目的mRNA的量和不同的引物有不同的退火和延伸溫度。
6、在進行5‘和3’RACEPCR的時候應該使用熱啟動。
7、重疊引物的設計會對全長的產生有幫助。另外,重疊的引物可以為PCR反應提供一個對照。並不是絕對的要利用設計的引物產生重疊片段。
8、引物GSP中的GC含量要在50-70%之間。這樣可以使用降落PCR。避免使用自身互補性的引物序列,否則會產生回折和形成分子內氫鍵。另外,避免使用與AP1互補的引物,尤其是在3‘末端。
9、如果要用重疊片段來檢測設計的引物,GSp1和GSp2之間至少是100-200鹼基。只有這樣才可以用擴增的產物來鑑定設計的引物是否正確。
10、降落PCR可以明顯的增加RACEPCR產物的特異性。在最開始的循環中,退火溫度高於AP1引物的Tm值,可以增加對特異性條帶的擴增。隨後的退火和延伸的溫度降回到AP1的溫度,可以進行隨後的PCR循環。
11、驗證基因特異性引物的對照:
單個引物的陰性對照:只用一個引物GSP來進行陰性對照。這樣不應該產生任何的條帶。如果可以看到明顯的產物,應該改變循環的參數,或重新設計原始引物。
利用兩個GSPS進行陽性對照:(只有兩個GSP可以產生重疊的時候才可以採用此步。)為了確定RNA樣品中目的基因確實表達,利用兩個GSP和接頭連線的cDNA來產生陽性對照。可以產生兩個引物之間的重疊大小的片段。如果沒有這個片段,應該重複cDNA的合成,或者從一個不同的組織或細胞來源進行cDNA的合成。
12、製備和抽提polyA+RNA不要使用DEPC處理過的水。純化完mRNA之後,利用瓊脂糖凝膠電泳檢測mRNA的質量。哺乳動物的mRNA樣品是0.5-12kb的拖帶,在其中有4.5和1.9kb的rRNA的條帶。非哺乳動物的mRNA應略小。
比較與最佳化
 RACE技術
RACE技術目的:研究環化法、末端脫氧核糖核酸轉移酶法和SMART反轉錄酶法3種常用5'RACE的技術的特點,並對它們進行了最佳化。
方法:以播娘蒿RNA為模板,先按文獻標準方法進行5'RACE,然後通過增加反應時間,提高部分反應物濃度等方法進行最佳化。
結果:未最佳化前環化法和TdT酶法條帶幾乎不可見,SMART反轉錄酶法隱約可見條帶,但不清晰。最佳化條件後,環化法出現彌散條帶,TdT酶法勝之,但條帶依舊彌散,而SMART反轉錄酶法最佳,獲得清晰特異性條帶。結論SMART5'RACE效果最好,其次為TdT酶法,環化法效果有待改進,同時通過最佳化條件可以明顯增加產物的特異性,為實驗研究中5'RACE方法的選擇提供了依據。
實驗步驟
cDNA第一條鏈的合成:
我們建議進行cDNA合成的對照反應,這樣可以對樣品的cDNA的合成進行鑑定。加入各種試劑之後,在氣浴中42度保溫一個小時。
注意:在水浴或酒精浴中保溫回減少反應體積,從而降低第一鏈的合成效率。將管放於冰上,以終止第一鏈的合成反應。直接進行第二鏈的合成。
cDNA第二鏈的合成:
第二鏈合成的酶混合物中,含有聚合酶、RNaseH和連線酶。T4DNA聚合酶的功能是補平dscDNA的末端。我們建議做陽性對照,試劑盒中提供人類骨骼肌的mRNA。
1、建議進行陽性對照,cDNA的質量取決於製備的polyA+RNA的質量。非哺乳動物樣品的mRNA大約在0.5-3kb之間。
2、通過電泳檢測cDNA的產量,與對照進行對比,這樣可以有利於在以後的步驟中對cDNA進行稀釋。
3、按照程式進行連線反應。
4、如果沒有對比樣品和對照的產量,利用Tricine-EDTAbuffer製備接頭連線的dscDNA的1/50和1/250的稀釋物,用兩種稀釋物進行RACEPCR反應,直到鑑定出哪一種稀釋可以得到好的效果。
PCR擴增
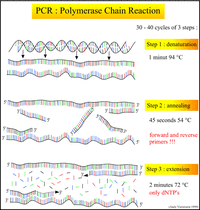 RACE技術
RACE技術進行5’和3’的RACE-PCR擴增。
利用以下的程式進行降落PCR反應:
94度30秒
5個循環:
(1)94度5秒
(2)72度4分
5個循環:
(1)94度5秒
(2)70度4分鐘
20-25個循環:
(1)94度5秒
(2)68度4分鐘
注意:
我們建議使用降落PCR反應,這就要求GSP的Tm值≥70度。
1、當循環結束時,利用1.2%瓊脂糖凝膠電泳分析每一個管中的產物5μl,使用適當的分子量marker。
2、可以根據你的基因的特異性來設計最理想的循環參數。如果看不到帶或者只有微弱的帶,在68度多加5個循環。最佳的延伸時間取決於擴增條帶的長度。如果片斷的長度在2-5kb的時候,經常使用4min,0.2-2kb的時候將延伸時間減到2-3min,對於5-10kb的條帶,延伸時間增加到10min。
目前遇到的問題及解決方法
實驗中發現,做RACE反應實驗實際操作中仍存在不少困難。因此,對RACE反應條件進行反覆摸索是十分必要的。這是因為:
1. 在5'-RACE包含了有3個連續的酶反應步驟(反轉錄、同聚物加尾和PCR擴增),每一步都可能導致失敗;
2. 擴增DNA末端的特異性完全依賴錨定引物及擴增DNA模板樣品的不均一性,因而特異性一般很低,常呈現不清晰的成片條帶或截短的產物背景。因此,使用RACE技術擴增得到的特異末端片段,所獲得的重組克隆最好能夠全部測序,以排除RACE實驗結果中擴增產物假陽性和假陰性,最終有可能獲得新基因的全序列。
隨著RACE的不斷改進和完善,最佳化條件下PCR擴增效率和忠實性的提高及PCR產物克隆技術的迅速發展,RACE必將在基因克隆及基因表達研究中發揮越來越大的作用。
RACE的另一種代表方法-Self-Ligation法
反轉錄(RT)反應------Hybrid RNA的分解------單鏈cDNA的自身連線------PCR擴增5'未知區域------目的DNA片段的切膠回收------DNA序列測定
克隆和測序
1、可以利用膠回收試劑盒來回收RACE產物,此試劑盒適合回收2.5kb以下的RACE產物;對於長的片段,可以通過電洗脫獲得好的結果。如果你選擇使用其他的純化方法,最後用Tricine-EDTAbuffer30μl重新懸浮DNA樣品。2、電泳5μl回收的樣品來鑑定回收的質量。
3、將回收的PCR產物直接克隆到/A型的PCR克隆載體中。另外還可以利用接頭和/或cDNA合成引物中的Not1、Srf1、Xma1、ECOR1等酶切位點,將產物克隆到常規載體中。
4、對於5‘端的RACE產物,我們建議挑取至少8-10個不同的克隆以獲得5‘端的最大可能性的序列。(反轉錄並不總是進行到mRNA模板的5’末端,尤其是長模板。另外,T4DNA聚合酶會移走5‘末端的0-20個鹼基。)
5、一旦鑑定了含有插入片斷的克隆,應該獲得多的序列信息。理想的是,可以對整個開放讀碼框進行測序。包括5‘和3‘的非翻譯區。
cDNA的獲得
 RACE技術
RACE技術通過部分或全部測序鑑定了RACE產物後,可以通過兩種選擇獲得全長的cDNA。通過PCR和克隆的方法。 通過PCR的方法獲得全長cDNA:
1、擴增長的cDNA需要較長的延伸時間,但是如果延伸的時間過長,可以產生拖帶,所以要慎重的設計引物。
2、根據從5‘和3‘RACE產物獲得序列信息設計5’和3‘GSP引物。這些引物應該來自cDNA的3’或者5‘的末端,應該是23-28nt長。不應該在引物的末端加上限制性位點,這樣會導致高背景。在某些時候可以設計3’和5‘的嵌套引物。但是還是應該先利用一對引物進行PCR反應。
3、進行如下的熱循環:
94度30秒
25個循環:
(1)94度5秒
(2)72度2-15分鐘
4、延伸的時間應該等於預期的cDNA長度加上2分鐘。例如:預期得到6kb的條帶,用6+2=8分鐘的延伸時間。注意:如果沒有條帶或者條帶弱,增加5個循環;或者最佳化PCR的條件。
5、在1.2%的瓊脂糖凝膠上分析5μl的樣品。通常情況下,可以見到一條單一帶,如果這樣,利用膠來純化全長的cDNA。
6、製備1.2%的TAEbuffer製備瓊脂糖凝膠。不使用TBEbuffer,TBE的膠很難製備全長的cDNA。
7、將剩下的45μl反應物點樣,選用適當的marker。
8、利用長波紫外觀察cDNA(≧300nm)切下全長的cDNA。注意:應該儘量減少紫外對cDNA的照射。
9、利用膠回收試劑盒回收cDNA。此試劑盒適合回收2.5kb以下的RACE產物;對於長的片段,可以通過電洗脫獲得好的結果。如果你選擇使用其他的純化方法,最後用Tricine-EDTAbuffer30μl重新懸浮DNA樣品。
10、將全長的cDNA克隆到T/A型的PCR克隆載體中。
通過克隆產生全長的cDNA:
1、如果你已經獲得了含有重疊部分的5‘和3’RACE產物,同時在cDNA的重疊部分含有一個酶切位點的話,通過克隆技術可以獲得全長的cDNA。
2、利用酶切獲得的3‘和5’擴增產物,並且利用T4DNA連線酶將它們連線起來。利用接頭和cDNA合成引物中的內切酶將最終的全長cDNA克隆到載體中。
3、在哺乳動物基因組中,NOT1和Srf1酶切非常稀少,大約106bp才出現一次,因此在絕大多數的cDNA中是不出現的。
